
内科学 第10版 「多発性筋炎・皮膚筋炎」の解説
多発性筋炎・皮膚筋炎(リウマチ性疾患)
多発性筋炎(polymyositis:PM)は,四肢近位筋,頸部筋の対称性筋力低下をきたし,骨格(横紋)筋の炎症,変性,再生を主病変とする,原因不明の特発性炎症性筋疾患(idiopathic inflammatory myopathy)の代表疾患である.筋炎症状に加え,Gottron徴候などの特徴的な皮膚症状を伴う場合には皮膚筋炎(dermatomyositis:DM)とよばれる.間質性肺炎,関節炎,消化管障害,心筋障害など筋以外の臓器障害も合併する.また,自己抗体が検出される全身性自己免疫疾患でもある.
分類
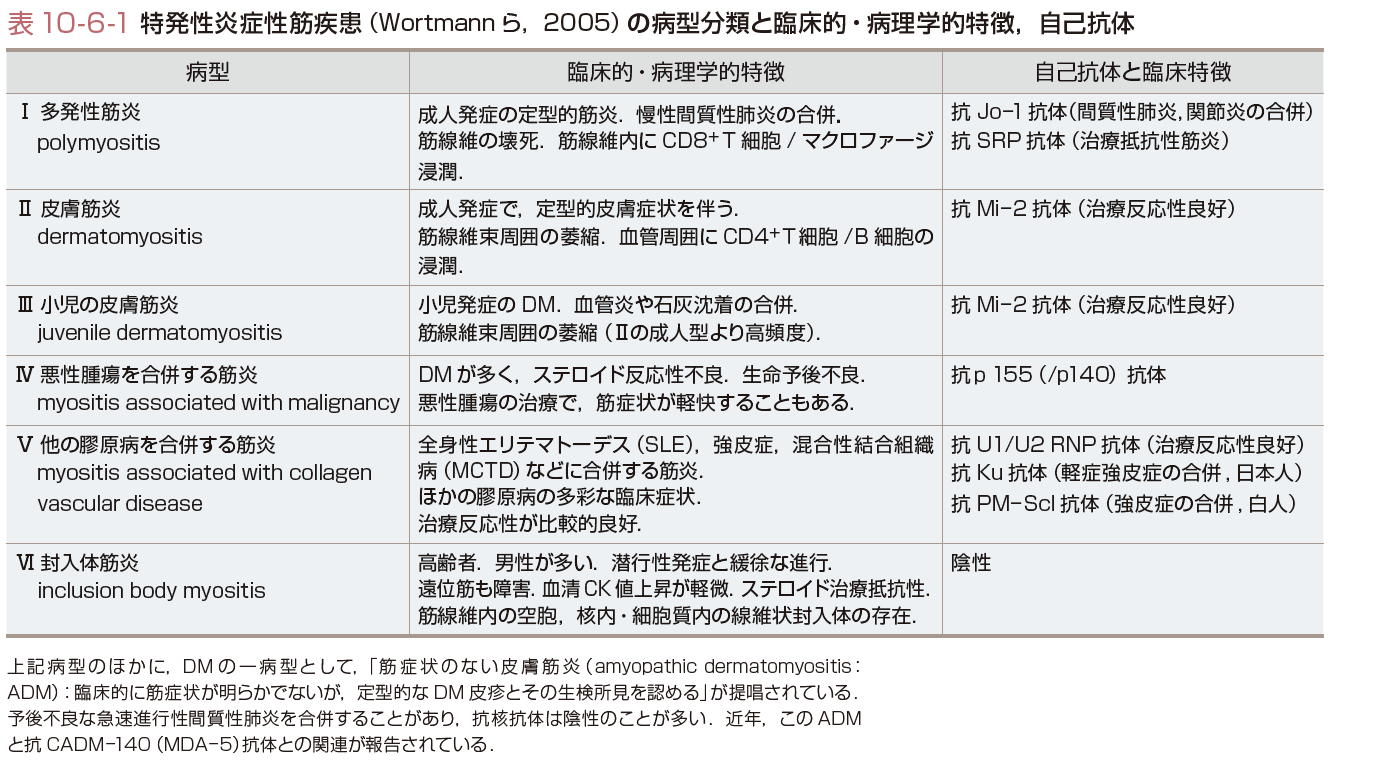
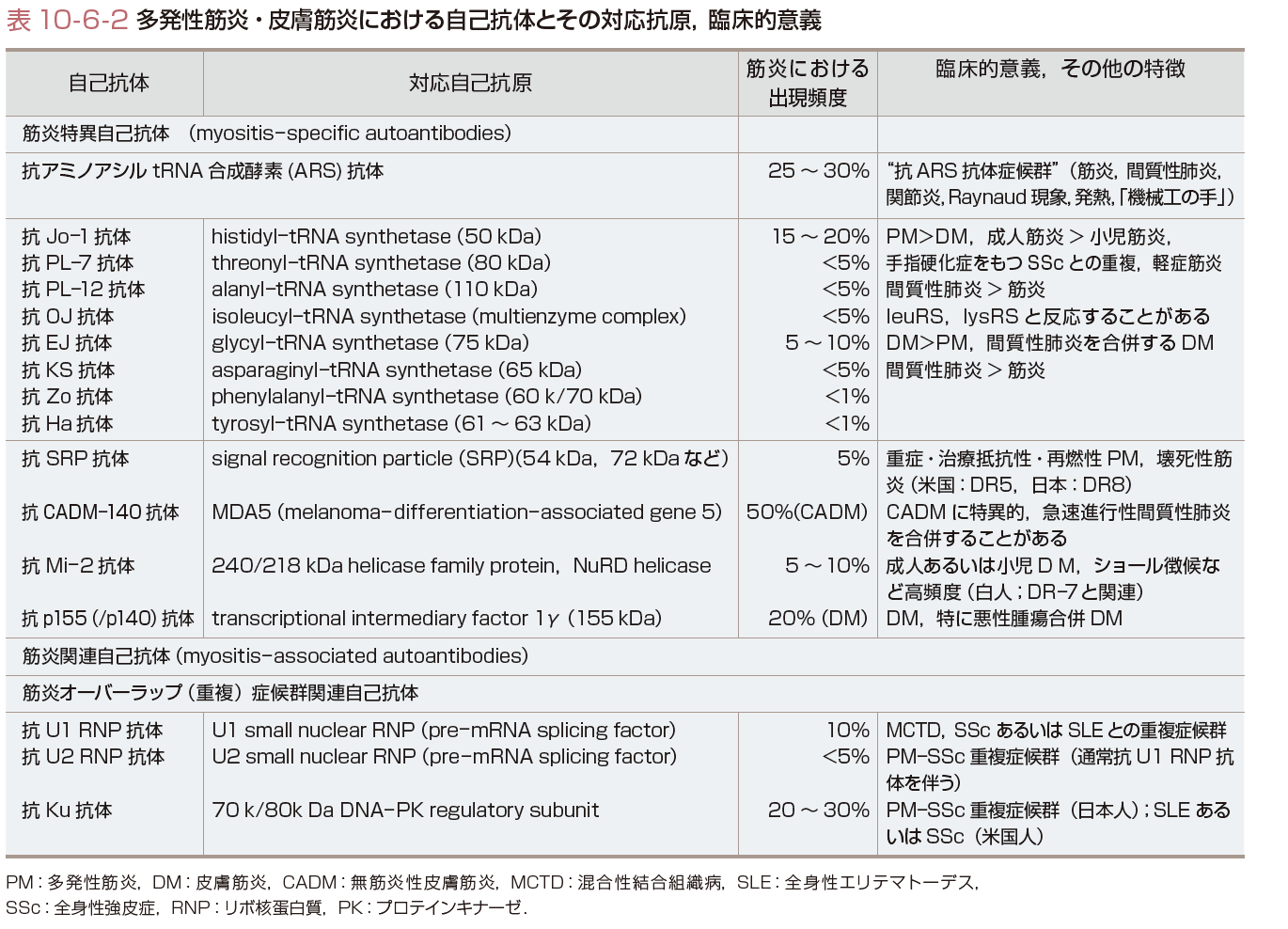
PM/DMの病像は多彩で,その病型分類は治療法の選択,予後の推定に有用である.その中で,発症様式,臨床経過,皮膚症状,悪性腫瘍合併の有無で分類する,Bohanの分類(Wortmannによる改訂分類)が広く用いられている(表10-6-1).筋炎に特異的な自己抗体(myositis-specific autoantibodies)が,臨床病態と関連することから,これらの自己抗体により病型分類することも臨床的に有用である(表10-6-1,10-6-2).
疫学
わが国の年間発病率は5〜10/100万人,有病率は2〜5/10万人である.成人では1:2の割合で女性に多いが,小児例では性差はない.発症年齢は,あらゆる年齢層に発症するが,小児期(5〜14歳)と成人期(35〜64歳)にピークをもつ二峰性分布を示す.
病因
原因はいまだ不明であるが,遺伝的素因(HLAなど)に,何らかの環境要因(感染性物質:ウイルス,細菌,寄生虫など,非感染性物質:食物,薬物,毒物,環境(暴露)物質など)が加わり,免疫異常が誘発され,筋炎を発症すると考えられている.この免疫異常は,PM/DMにおける筋組織への免疫担当細胞浸潤,ほかの自己免疫疾患の合併,および筋炎に特異的な自己抗体の存在などより,自己免疫異常が関与するとされる.
自己免疫異常と発症機構
1)細胞性免疫異常:
PMや封入体筋炎では,筋組織に浸潤する炎症性細胞は,筋線維内部や周辺には細胞傷害性(CD8+)T細胞が主体で,T細胞の浸潤した筋線維ではMHCクラスⅠ抗原の発現が増強し,筋線維膜上には接着分子ICAM-1の発現が認められる.これらの所見は,PMや封入体筋炎では筋線維表面抗原を標的とする細胞傷害性T細胞が筋傷害に関与していることを示している.
2)液性免疫異常:
DMでは血管周囲へ(CD4+)T細胞とB細胞が優位に浸潤し,C5-9の補体後期成分が膜傷害性複合体として毛細血管,小動脈壁に沈着することから,皮膚・筋組織の毛細血管を標的とする液性免疫の関与が考えられている. ①遺伝的素因をもつヒトに筋細胞構成蛋白と分子相同性を有するウイルスなどの環境因子が作用し,自己抗体が産生される,②アポトーシスの結果,アミノアシルtRNA合成酵素(aminoacyl tRNA synthetase:ARS)に対する自己免疫が誘導される,③ARS自身が化学誘引により炎症細胞浸潤を誘導し,筋組織傷害を引き起こす,などの可能性も報告されている.
病理
筋線維の大小不同,変性と壊死・再生像,血管周囲や間質へのリンパ球を主体(マクロファージや形質細胞も混在)とする炎症性細胞浸潤,間質の線維化を特徴的所見として認める(図10-6-1A).炎症性細胞は,PMではCD8+T細胞が筋内膜(endomysium)周辺に,DMではCD4+T細胞やB細胞が筋周膜(perimysium)と血管周囲に浸潤するのが特徴とされる.また,DMでは筋束周辺の筋線維萎縮(perifascicular atrophy)がよくみられる.小児DMでは,血管炎による血栓の形成や梗塞像を認めることもある.
DMの皮膚生検組織では,表皮の萎縮と基底層の液状変性,血管拡張,炎症性細胞浸潤が認められる.
臨床症状(図10-6-1)
PM/DMの臨床症状は筋症状と筋以外の症状(筋外症状)に分けられ,多彩である.初発症状では筋力低下(特に下肢)(50〜70%),皮膚症状(30〜40%),関節痛,筋痛(10〜20%)が多い.その他,発熱,全身倦怠感,体重減少などの全身症状,Raynaud現象などで発症する.
1)筋症状:
筋力低下は本症の 主症状で,好発部位は四肢近位筋群(下肢95%,上肢75%),頸部屈筋群(70%),および咽頭・喉頭筋群(70%)である(図10-6-1).筋力低下は亜急性型,慢性型では下肢筋から徐々に始まり,慢性経過をとる症例もある.下肢帯筋力の低下により,しゃがみ立ち,階段の昇降,入浴が困難となり,歩行障害も出現する.上肢帯筋力も失われ,髪をとかすなどの,腕の挙上・保持などが障害される.頸部屈筋群の障害により仰臥位からの頭部挙上が困難となったり,咽頭・喉頭筋群の傷害で嚥下障害,構語障害(鼻声)も生じる.重症例では呼吸筋(横隔膜・肋間筋)傷害による呼吸不全を起こすこともまれにある.筋痛もしばしば認める.筋萎縮は慢性例に特徴的で,筋ジストロフィとは異なり,病初期に認めることはまれである.
2)筋以外の臨床症状(筋外症状):
a)皮膚症状:約30〜40%の症例は特徴的な皮疹を呈し,DMと診断される.皮膚症状は顔面と四肢関節背側面に好発する.ヘリオトロープ疹(heliotrope rash)は上眼瞼部の浮腫性の紫紅色の皮疹で,日光暴露で増悪する(図10-6-1B).Gottron徴候(Gottron’s sign)は手指関節背側面の落屑性紅斑で,DMに特異的である(図10-6-1C).その他,肘,膝,内果など大小関節背側面,前頸部-上胸部(V徴候:V-sign),肩・上背部(ショール徴候:shawl sign)の落屑性紅斑もみられる.爪周囲紅斑,爪床部の毛細血管拡張,梗塞はDMに特異的でないが,高頻度にみられる.抗Jo-1抗体などの抗ARS抗体陽性例で手指先端指腹の裂溝を伴う角質化と色素沈着(機械工の手(mechanic’s hand))がみられる.小児例で血管炎による皮下の多発性石灰沈着を認めることがあるが,成人例では少ない.Raynaud現象は,約30%の症例にみられるが,軽症で,強皮症のように皮膚潰瘍,手指壊疽を伴うことは少ない.DMの皮膚症状のみで,筋炎所見の明らかでない症例は,無筋症性皮膚筋炎(amyopathic dermatomyositis:ADM)とよばれ,急速進行性間質性肺炎の合併に注意を要する.
b)関節症状:多発関節痛・関節炎も約30%に認めるが,軽症,一過性で骨の破壊・変形を残さないことが多い.まれであるが,抗Jo-1抗体陽性例で亜脱臼などの変形を伴う慢性関節症が報告されている. c)呼吸器病変:間質性肺炎が約40〜50%の症例に認められ,重要な生命予後因子である(図10-6-1D).自覚症状(乾性咳,労作時呼吸困難),身体所見(下肺野を中心とする捻髪音),肺機能検査所見(拘束性障害,肺拡散能の低下)は,特発性間質性肺炎と類似するが,無症状の例も少なくない.間質性肺炎は筋炎症状とともに発症することもあるが,先行することも,後から合併することもある.多くの症例が慢性に経過し,病理組織学的所見は非特異性間質性肺炎(NSIP),器質化肺炎(OP),通常型間質性肺炎(UIP)を示す.しかし,大量ステロイド療法に反応せず,呼吸器症状発現後数カ月で死亡する急速進行性間質性肺炎はきわめて予後不良な病態で,ADMに多く,病理組織学的にはびまん性肺胞傷害(DAD)を呈する.重症間質性肺炎例には縦隔気腫や皮下気腫が合併することがあり,DM例に多いとされる.その他の肺病変では,①食道運動障害による嚥下性肺炎,②呼吸筋筋力低下による低換気と分泌物貯留,③投与薬剤による日和見感染,④薬剤性肺臓炎(③④は合併症)などがある. d)心病変:心電図異常(ブロック,期外収縮,ST-T変化など),心筋傷害による心不全などを認めることがある.心臓超音波検査で心膜炎も認めるが,無症候性のことが多い.心病変は生命予後因子として,注意を要する. e)消化器病変:咽頭筋力低下,食道の蠕動低下による嚥下障害が約30%にみられ,一般に重症例に多い.まれに,小腸,大腸の蠕動低下も認める.小児DMで血管炎による消化管の潰瘍を合併し,腹痛,下血をきたす例もある. f)悪性腫瘍:PM/DMの7〜30%に悪性腫瘍が合併し,生命予後と関連するため,その検索は重要である.特に,DM(高齢,男性)と密接に関連し,診断後1〜2年以内に発症することが多い.あらゆる悪性腫瘍を合併し,胃,肺,前立腺,子宮,卵巣,乳腺,大腸癌,さらにリンパ腫も報告されている.悪性腫瘍の切除,あるいは治療により,筋症状が改善する例もあり,病因的にも注目される.
検査成績
1)筋炎診断のための検査:
a)血液・尿検査:血清クレアチンキナーゼ(CK),アルドラーゼ,LDH,ASTなどの筋原性酵素が筋傷害の程度と範囲を反映して上昇する.CKアイソザイムはMM型を主体とする.これらは,筋炎の診断ばかりでなく,筋炎の活動性を示し,治療効果判定の重要な指標となる.また,筋傷害が高度の場合,ミオグロビン血症,ミオグロビン尿症を認め,尿細管障害や腎不全の合併に注意する.
b)免疫血清学的検査:PM/DMに特異的に見出される自己抗体(myositis-specific autoantibodies)は,診断,病型の分類,予後の推定,治療法の決定など臨床的に有用である(表10-6-1,10-6-2).抗Jo-1抗体は,PM/DMの20〜25%と最も高頻度に検出される,疾患標識自己抗体である.本抗体陽性例は筋炎のほかに,間質性肺炎,多発性関節炎,Raynaud現象,機械工の手などを高頻度に合併する「抗ARS抗体症候群」とよばれる一病型を形成する.抗シグナル認識粒子(signal recognition particle:SRP)抗体は,PM(とくに定型的成人PM)に低頻度(<5%)ながら特異的に検出される.ステロイド抵抗性の難治性筋炎・壊死性筋症と関連する.抗Mi-2抗体は,DMの疾患標識自己抗体である.本抗体陽性例は,間質性肺炎の合併が低頻度で,ステロイド反応性良好な病型と関連する(表10-6-2). c)筋電図(electromyography:EMG):筋電図と神経伝導速度の測定は,PM/DMの診断および神経疾患による筋病変との鑑別に有用である.炎症性筋疾患をもつ80〜90%の症例が ,「筋炎の3徴」①筋原性所見(随意収縮運動時:神経筋単位電位の持続時間の短縮,低電位,多相性),②筋内神経線維の障害を示す脱神経電位(安静時:線維自発電位(fibrillation)や棘波),③複合反復電位,を示す(表10-6-3A).
d)核磁気共鳴(magnetic resonance imaging:MRI)検査:筋肉MRIは,非侵襲的で,筋炎の活動性や病変(炎症,萎縮,脂肪変性など)の評価,筋生検部位の決定,経時的観察に有用である.MRIのT2強調画像/脂肪抑制画像で,筋炎の炎症性変化部位が高信号領域,T1強調画像で正常,脂肪変性はT1,T2両強調像で高信号に描出される. e)筋生検:本検査の結果,特徴的筋病理組織所見(図10-6-1A)を認めると確定診断される.筋生検の部位は,MRIで異常を認めた筋,あるいはEMGで異常のあった同名反対側の筋(三角筋,上腕二頭筋,大腿四頭筋など)を選択し,ステロイド治療開始前に行うべきである.
診断
PM/DMの診断は臨床症状(徒手筋力テストによる筋力低下の判定,特徴的な皮膚症状)および検査成績(血清筋原性酵素値・自己抗体,筋電図,筋生検所見)を組み合わせ,総合的に診断する.従来から広く用いられているBohanとPeterによる診断基準(表10-6-3A)と厚生省自己免疫疾患調査研究班の改訂診断基準(表10-6-3B)を示す.
鑑別診断
筋力低下や筋痛をきたす疾患がおもな鑑別疾患となる.神経筋疾患(遺伝性筋疾患,重症筋無力症など),内分泌(甲状腺機能低下,ステロイド筋症)・代謝性疾患(周期性四肢麻痺,糖原病),電解質異常,感染症(インフルエンザ,肝炎,コクサッキー ウイルス),薬剤(d-ペニシラミン,スタチン系抗脂質異常症薬)の影響,サルコイドーシス,ほかのリウマチ性疾患,悪性腫瘍による腫瘍随伴症候群,などが大切である.筋ジストロフィと筋外症状,炎症所見,免疫学的異常などが乏しいPMとの鑑別が困難なことがあり,発症年齢,家族歴,遺伝子検査,筋組織所見などにより鑑別する.
治療
治療の目標は,筋炎の鎮静化,筋力の回復,日常生活活動の向上と,合併する臓器障害の改善にある.血清CKなどの筋原性酵素は筋炎の活動性指標となり,治療効果の判定に有用である.しかし,治療抵抗性筋炎,急速進行性間質性肺炎など難治性病態に対する治療法は確立されていない.
1)筋炎の治療:
a)一般療法(日常生活上の注意):急性期・亜急性期には安静を保つ.慢性期には筋の拘縮に注意する.食事は高蛋白,高カロリー食とする.外傷,感染,薬物などは増悪因子となり,避けるよう指導する.特に,DMでは皮疹を増悪させる日光(紫外線)暴露を避けるように注意する. b)リハビリテーション:発熱などの全身症状や筋症状の強い急性期は安静を原則とする.関節拘縮を予防するため,関節の屈曲・伸展運動を1日数回行い,関節は良肢位に保つ.長期間の安静は筋萎縮(disuse atrophy)をきたし,筋力回復を遅らせるため,筋炎鎮静化(血清CK値が正常化)後は,筋力回復のための理学療法を開始する.安静期間は通常2〜4週間である.運動量は,筋力低下の程度,血清筋原性酵素値を指標に徐々に増やす.
c)薬物療法:
ⅰ)副腎皮質ステロイド:副腎皮質ステロイドが,炎症性筋疾患の第一選択薬として経験的に用いられ,その有効性が認められている.成人発症の典型的PM/DMにはプレドニゾロン(PSL)1.0〜1.5 mg/kg /日(PSL 40〜60 mg/日)を初回投与量とする(小児発症DMでは1〜2 mg/kg/日).2〜4週間継続後,筋力の改善とCK値を指標として1〜2週ごとに10%の割合で漸減する.血清筋原性酵素値の低下は,一般に筋力の改善に先行する.PSL 15 mg/日以下で再燃することがあり,以後の減量は慎重に行う.PSL 5〜10 mg/日を維持量として長期投与(1年以上)を続ける.治療抵抗症例,血管炎を伴う重症小児DMではステロイドパルス療法(メチルプレドニゾロン1000 mg/日を3日間点滴静注)も選択される.特に,筋原性酵素値の上昇のない筋力低下を認める場合,ステロイド筋症の鑑別が必要となる.治療は,ステロイドを速やかに減量し,PSL 30 mg/日以下にする.
ⅱ)免疫抑制薬:標準的な大量ステロイド療法を施行後も,血清CK値の改善,筋力の回復を認めないステロイド療法抵抗症例,ステロイドの副作用のため減量・中止が必要な症例では,免疫抑制薬が併用される.免疫抑制薬としては,メトトレキサート,アザチオプリン,シクロホスファミド(間欠大量静注療法も含む),シクロスポリン,タクロリムスなどが用いられる.近年,抗ARS抗体陽性例の筋炎や再燃性間質性肺炎に対するタクロリムスの有効例が報告されているが,現在のところ,保険承認薬の適用外使用であり,その投与にあたっては患者へのインフォームドコンセントが必要である. ⅲ)ガンマグロブリン大量静注(intravenous high-dose immunogloburin therapy:IVIG)療法:ガンマグロブリン1 g/kg/日を毎月2日間,あるいは400 mg/kg/日を毎月5日間を投与し,ステロイド抵抗性の難治性筋炎,再燃性筋炎,封入体筋炎での有効性が報告されている.副作用が比較的少なく,有効な治療法として期待されるが,長期有効性が確認されていないため,その適応は慎重な検討が必要である.
臨床経過・予後
大多数(75%)例で筋炎(筋症状)に対するステロイド療法の効果を認め,日常生活が可能となる.しかし,診断がDMよりPM,高度な筋力低下,診断や治療までの期間が長いこと,高齢発症などが,筋症状の機能予後の不良因子とされている. 生命予後は,悪性腫瘍の合併のないものは比較的良好で,5年生存率90%,10年生存率80%とされる.生命予後は,悪性腫瘍合併,間質性肺炎(特に急性間質性肺炎),感染症,心肺合併症(誤嚥性肺炎,呼吸筋傷害による呼吸不全,臨床的に明らかな心病変)により左右される.[平形道人]
■文献
平形道人:多発性筋炎・皮膚筋炎.リウマチ病学テキスト(日本リウマチ学会 日本リウマチ財団編),pp222-234, 診断と治療社,東京,2010.
Miller FW: Inflammatory myopathies: Polymyositis, dermatomyositis, and related conditions. In: Arthritis and Allied Conditons, 15th ed (Koopman WJ, Moreland ML ed), Lippincott Williams & Wilkins, Baltimore, pp1593-1620, 2005.
Wortmann RL: Inflammatory diseases of muscle and other myopathies. In: Kelly’s Textbook of Rheumatologys. 7th ed (Harris ED, Budd RC, et al ed), pp1309-1335, Elsevia Saunders, Atlanta, 2005.


出典 内科学 第10版内科学 第10版について 情報
Sponsored by ![]()