
内科学 第10版 「心肥大と拡張」の解説
心肥大と拡張(心血管代謝と機能)
(1)心肥大形成の分子メカニズム
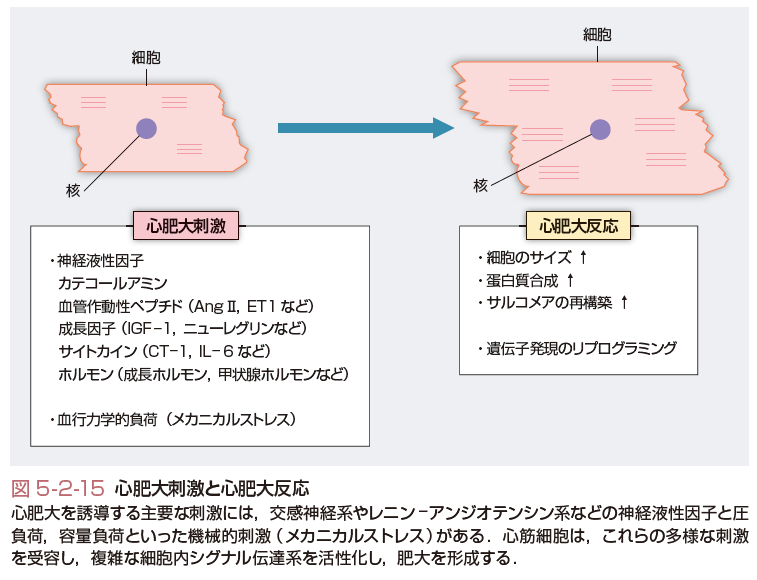
心肥大を誘導する主要な刺激には,神経液性因子とバイオメカニカルな機械的刺激(メカニカルストレス)がある(図5-2-15).カテコールアミンや,アンジオテンシンⅡ(Ang Ⅱ)やエンドセリンなどの血管作動性ペプチド,成長因子,サイトカイン,ホルモンなどの神経液性因子は,心筋細胞に発現するそれぞれの受容体,つまりG蛋白質共役型受容体や受容体チロシンキナーゼ,gp130と複合体を形成するサイトカイン受容体などと特異的に結合し活性化する.これらの多様な受容体の活性化をトリガーとして,複雑な細胞内シグナル伝達系が惹起され,心肥大反応が誘導される.特に心肥大反応の誘導に重要なシグナル伝達系として,mitogen-activated protein kinase (MAPK)系やカルシニューリン-nuclear factor of activated T cells (NFAT)系,phosphatidylinositol 3-kinase (PI3K)-Akt-mammalian target of rapamycin (mTOR)系,Janus kinase (JAK)-signal transducers and activators of transcription (STAT)系があげられる(Heinekeら,2006).また,cyclin-dependent kinase-7 (CDK7)やCDK9による転写伸長反応の制御やさまざまなリン酸化酵素によるclass Ⅱ histone deacetylase (HDAC)の機能修飾も,心肥大反応の誘導に深く関与している(Heinekeら,2006).
一方,メカニカルストレスを受容する心筋細胞のメカノセンサーとして,細胞と細胞外マトリクスとの接着を担うインテグリンを中心とした分子複合体とサルコメアのZ線を構成する分子複合体が重要である.特に心筋細胞では,インテグリンとZ線とはコスタメアという蛋白質複合体によって構造的に連結されている.特に,コスタメアに存在するインテグリン結合キナーゼやmelusin,Z線に存在するmuscle LIM protein (MLP)はメカニカルストレスに対する心機能の代償機構に重要な役割を果たしていることがノックアウトマウスや変異ゼブラフィッシュを用いた解析によって明らかとなっている.しかし,これらのメカノセンサーが心肥大形成にどのように関与しているか,またその詳細な分子機構については不明な点が多く残されている. また,細胞膜上の伸展感受性イオンチャネルやG蛋白質共役型受容体であるAng Ⅱタイプ1(AT1)受容体もメカニカルストレスにより活性化する.特に,メカニカルストレスによるAT1受容体の活性化はAng Ⅱ非依存的に生じ,特異的な構造変化によりG蛋白質やJAK2を介して心肥大形成を誘導することが報告され,最近大きな注目を集めている.
(2)求心性心肥大と遠心性心肥大
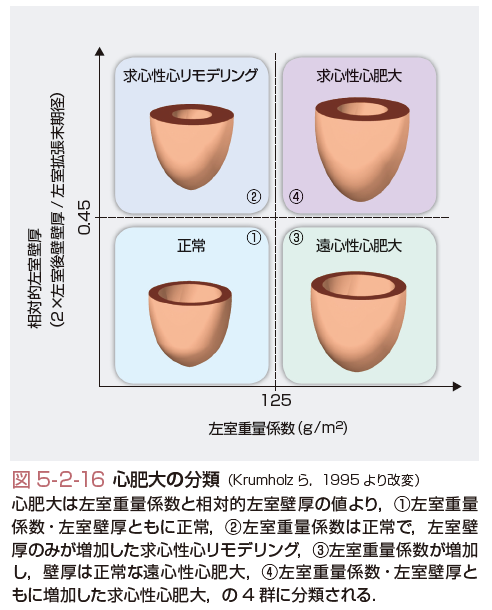
心肥大はその形態により図5-2-16に示すように分類される.つまり,左室重量係数(左室重量 g/体表面積 m2)と相対的左室壁厚(2×左室後壁壁厚 mm/左室拡張末期径 mm)の値より,①左室重量係数・左室壁厚ともに正常,②左室重量係数は正常で,左室壁厚のみが増加した求心性心リモデリング,③左室重量係数が増加し,壁厚は正常な遠心性心肥大,④左室重量係数・左室壁厚ともに増加した求心性心肥大,の4群に分類される(Krumholzら,1995).一般的に,高血圧や大動脈弁狭窄症などの圧負荷が増大する病態において求心性心肥大が生じ,僧帽弁閉鎖不全症や大動脈弁閉鎖不全症などの容量負荷が増大する病態において遠心性心肥大が生じる. 左室の内腔拡大などのマクロレベルでの構築の変化は,心拍出量が心臓の拡張末期容積に比例して増加するというFrank-Starling機構という点からは心機能維持にとって代償的であるが,その効果は十分ではなく,むしろ壁ストレスを増大させて,最終的には神経・液性因子の活性化状態とは無関係に心不全を進行させる.このような構築変化には,細胞外マトリクスの合成低下と分解亢進による量的変化,さらにコラーゲンのサブタイプの変換などの質的変化が深く関与していると考えられている.
(3)心不全への移行
過剰な血行力学的負荷が慢性的に持続すると,心肥大における代償機構が破綻し心不全に移行する.詳細な機序は明らかではないが,過剰な心筋細胞肥大に伴う酸素消費量の増加と,間質および血管周囲の線維化による酸素の拡散障害によって生じる心筋虚血が一因となっている.また,生理的心肥大や初期の代償性心肥大では毛細血管の増生を伴うために心筋細胞への酸素供給が保たれているが,非代償期には毛細血管新生が相対的に不十分となるために,心筋虚血がさらに悪化すると考えられている.このような心筋細胞の相対的虚血は,エネルギー平衡の破綻をきたして収縮能を低下させるとともに,心筋の細胞死や変性脱落により収縮障害を悪化させる.そのほかに,細胞レベルでは,筋小胞体の機能障害や収縮蛋白質の生合成の低下なども収縮障害を進行させる.[小室一成・赤澤 宏]
■文献
Heineke J, Molkentin JD: Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol, 7: 589-600, 2006.
Krumholz HM, Larson M, et al: Prognosis of left ventricular geometric patterns in the Framingham Heart Study. J Am Coll Cardiol, 25: 879-884, 1995.
Levy D, Garrison RJ, et al: Prognostic implications of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med, 322: 1561-1566, 1990.
出典 内科学 第10版内科学 第10版について 情報
Sponserd by ![]()