
内科学 第10版 の解説
自己免疫.リウマチ性疾患と膠原病(リウマチ性疾患総論)
膠原病(collagen disease)は1942年Klempererにより提唱された概念であり,全身性エリテマトーデス(SLE)や強皮症の病理学的研究をもとに,結合組織全般に変化がみられ細胞外成分に特徴の認められる急性および慢性の疾患とした.それまでの疾患は,肝臓,肺,心臓など,各臓器に主病変があると考えられていたので,新しい概念の提唱であった.
膠原病とは単一の疾患を示す臨床診断名ではなく,病因を意味する用語でもない.結合組織がおもな病変であるので,結合組織病ということもある(欧米では現在はconnective tissue diseaseという).一般に結合組織と血管を主病変とし,自己抗体を高頻度に伴う多臓器性の非腫瘍性,非感染性の慢性難治性疾患と考えてよい.
病理組織学的には,全身の膠原線維にフィブリノイド変性という共通の病変がみられる.一方,膠原病は臨床的にはリウマチ性疾患(rheumatic disease)の範疇に入る.リウマチ性疾患とは,関節・筋肉・骨・靱帯・腱などの運動器の疼痛とこわばりを有する疾患のことである.ただしリウマチ性疾患には,変形性関節症,痛風など膠原病以外の疾患も含まれる.また一方,病因論的には膠原病は自己免疫疾患(autoimmune disease)と考えられている.ただし,自己免疫性疾患は,膠原病以外の甲状腺炎など臓器特異的自己免疫疾患があり,膠原病はこれらとは異なり全身性自己免疫疾患とされている.免疫応答と病態の関係で,後述のように両者の相違がいくつかあるが,これらは明確に区別されているわけではない.すなわち膠原病に関しては,病理学的,臨床的,病因論的にそれぞれの名称がある.
(2)自己免疫疾患と自己免疫
自己免疫疾患を大きく分けると,免疫反応の標的抗原と組織傷害が1つの臓器に限局している臓器特異的自己免疫疾患と,生体に広く分布している抗原,たとえば核内抗原に対しての免疫反応が主として観察され,多臓器にわたり傷害がみられる全身性自己免疫疾患とがある.膠原病は全体としてみると後者の全身性自己免疫疾患の範疇に入る.臓器特異的自己免疫疾患は,本来トレランス(免疫寛容)になっているはずの特定の臓器にだけ発現している自己の抗原に対してそのトレランスが破綻し,その自己抗原に対して免疫系が積極的に反応した結果であると考えられる.すなわちこの場合,問題となっている自己抗原に特異的な免疫反応が重要な役割を果たしていることは明らかである.
一方,膠原病がこのような免疫反応の延長線上にあるか否かはよくわかっていない.膠原病で検出される自己抗体の標的はほとんどが核内物質や細胞質分子なので,自己抗体自体が直接臓器傷害を起こしているとは考えにくい.すなわち,全身性自己免疫疾患では,いまのところ自己抗原に対する免疫異常と種々の病態とが一元的に理解できていない.
自己免疫現象とは,抗体またはT細胞が自己の抗原と反応する現象である.そして,自己免疫現象の結果引き起こされるのが自己免疫疾患である.ところが,健常人にもこれらの自己反応性の抗体やT細胞が存在することがわかっている.生体には自己との免疫応答を抑制するさまざまなメカニズムがあるが,それらが破綻すると自己免疫疾患が引き起こされる.したがって健常人に存在する自己免疫現象とそれぞれの疾病における自己免疫現象の質的,量的相違を的確に把握し,その原因,病態への関与,修復の方策を探ることが重要である.
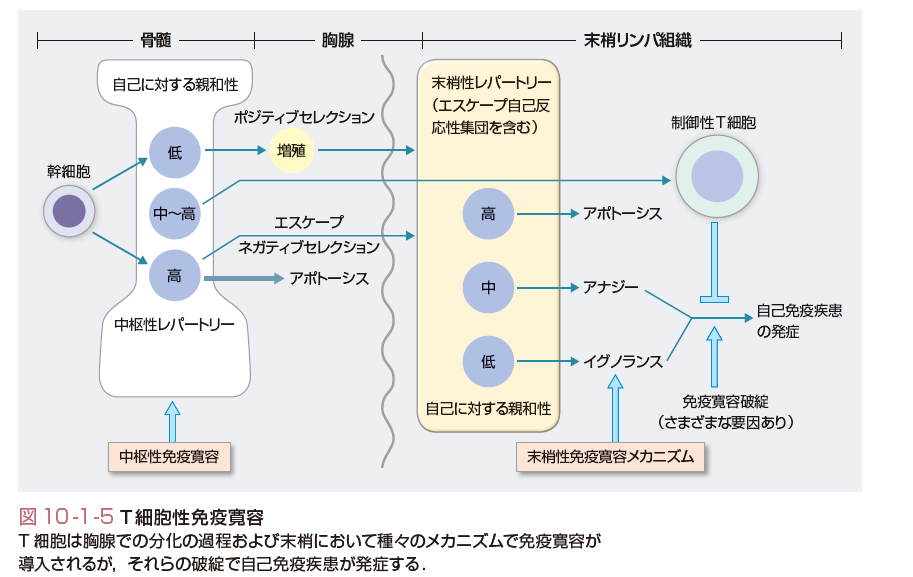
自己の抗原に対して免疫応答を生じない状況を自己免疫寛容(self-tolerance)とよぶ.獲得免疫応答の中心であるT細胞の場合,それが分化する胸腺内で十分量の自己抗原に暴露されると,アポトーシスによりクローン除去を起こす.これが中枢性免疫寛容である.しかし,このメカニズムにも限界があり,すべての自己抗原が胸腺で発現してはいないことや,自己抗原との反応性はあるがそれほど反応が強くないT細胞は末梢組織に出ていく.しかし,それでも自己免疫反応が顕性にならないのは,末梢での免疫寛容のシステムがあるからであり,これにはアナジー(anergy,クローン麻痺),クローン無視,制御性T細胞など種々のメカニズムがある.これらの異常で自己免疫現象と自己免疫疾患が引き起こされるのが主要因と考えられている.
(3)中枢性免疫寛容とその破綻
自己反応性のB細胞は正常のB細胞レパートリーのなかに存在する.そのいくらかは“natural autoantibodies”を産生する.これらは通常,IgMで低アフィニティ,自己抗原に対してpolyreactiveであり,初期生体防御や自己抗原のクリアランスに役立っており,自己免疫を防止する役割があるともいわれている.病態形成性のB細胞も正常のレパートリーのなかに存在するが,これらが活性化されるにはT細胞のヘルプを必要とする.これらを考慮すると,自己免疫疾患に至る免疫寛容の破綻の重要なポイントの1つはT細胞の免疫寛容の破綻にあると考えてもよい. T細胞の中枢性免疫寛容の異常で自己免疫疾患になる例は多くはない.自己免疫性多発内分泌症候群Ⅰ型(APECED)は,特発性Addison病に特発性副甲状腺機能低下症などの自己免疫性内分泌疾患と皮膚カンジダ症を高率に伴う症候群であるが,この原因遺伝子としてAIRE(autoimmune regulator gene)が同定された.そして,このAIRE遺伝子は臓器特異抗原の一部が胸腺で発現する現象に関与していることが判明した.すなわちこの遺伝子異常により臓器特異的抗原に反応するT細胞が胸腺で除去されないことで自己免疫疾患になると考えられている.
(4)末梢性免疫寛容の種々のメカニズム
大部分の免疫寛容の破綻はT細胞の末梢性免疫寛容の破綻によると考えられている(図10-1-5).末梢性免疫寛容の異常には,多くのメカニズムが示唆されているが,その代表的なものとして,まず隔絶抗原の免疫系への暴露がある.これは解剖学的構造より,免疫系に提示されていない自己抗原が,たとえば外傷や炎症などにより免疫系に暴露される現象である.外傷後の交換性眼炎や精管切断後の精巣炎などの例がある.また自己抗原の修飾も寛容の破綻の原因になり得る.たとえば自己抗原がほかの分子と複合体を形成する場合,抗原提示細胞内で通常と異なるプロセシングを受ける可能性があり,本来ほとんど提示されていなかったcryptic(隠れたの意味)なエピトープが抗原提示されることがあると考えられる.また蛋白質は翻訳後にアセチル化,脱アミド化,脱イミド化など多くの修飾を受けるが,この異常が生じると生体にとって新しい自己抗原が出現することになる.実際,関節リウマチでは脱イミド反応によりアルギニンがシトルリンに変換された自己抗原に対する自己抗体が特異的に生じていることが明らかとなりつつある.
分子相同性とは自己抗原と外来抗原の間に交差反応性があることをいう.A群連鎖球菌感染後のリウマチ熱は心炎や関節炎などの症状を呈するが,これはA群連鎖球菌のM蛋白と心筋のミオシンとの交差反応ではないかと考えられている.その他の末梢性免疫寛容の異常としては,制御性T細胞の異常,副刺激の異常発現,抑制性分子の異常,アポトーシスの異常,スーパー抗原など種々の可能性が報告されている.以下にそのおもなものとしてT細胞のアナジーと制御性T細胞について概説する.
a.T細胞のアナジー,機能低下のメカニズム
T細胞のアナジーは末梢のトレランスに重要な働きをしていると考えられるが,最近この分子メカニズムが少しずつ明らかにされている.アナジーとは抗原に出会ったときの,インターロイキン-2(IL-2)産生と増殖の両者の抑制と考えることができる.持続的な抗原刺激による生体内でのアナジー誘導には,CTLA-4という抑制性分子の発現増大が一部かかわっていると考えられる.
またアナジーとの関係ではE3ユビキチンリガーゼも注目されている.ヒトゲノムには1000種類ものユビキチンリガーゼE3があると想定されているが,いくつかの分子が抗原刺激からのシグナルを阻害することで免疫抑制に働いていることが明らかにされつつある.c-Cblは最初にアナジーとの関係で報告されたE3ユビキチンリガーゼであり,そのファミリーのCbl-bは,アナジーのT細胞に再刺激が入ったときに,PLC-γやPKC-θの発現抑制に働く.おそらく1つのE3ユビキチンリガーゼではなく,複数のE3ユビキチンリガーゼが共同でアナジーの誘導と維持に関与しているのではないかと考えられている.
b.種々の制御性T細胞
末梢でのトレランスに制御性T細胞が重要であることは明らかであるが,制御性T細胞にもいくつかの種類があり,それが少しずつ分類されてきている.自然発生制御性T細胞(naturally occurring regulatory T cell)の主要なものはCD4+CD25+でGITR+,CTLA-4+の細胞であり,おそらく自己抗原を認識して細胞接触によりエフェクターリンパ球を抑制する.さらに最近,これ以外の自然発生制御性T細胞も見いだされている.自然発生以外のものは誘導性制御性T細胞(induced regulatory T cell)とされており,種々のものが記載されている.Tr1やTh3とよばれるT細胞はそれぞれ,IL-10やTGF-βの存在下に分化してくる.
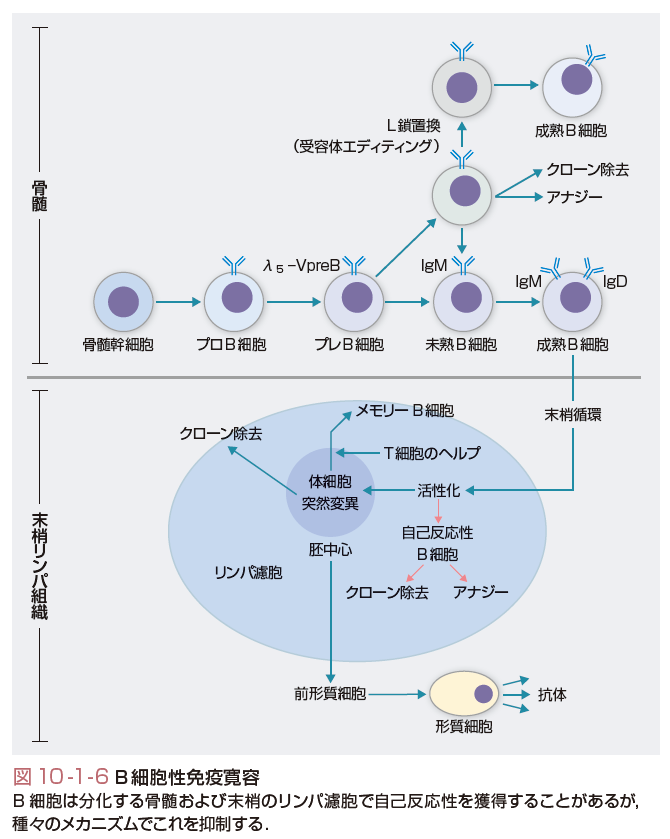
(5)B細胞と自己免疫,免疫寛容
最近,臨床での治療経験などの蓄積から,自己免疫におけるB細胞の役割が再び注目されている.すなわちB細胞を標的とした自己免疫疾患の治療が予想外の効果を発揮していることから,従来考えられていたB細胞の役割以上に免疫寛容,免疫調節に対するB細胞の役割があると推測されるようになっている(図10-1-6).たとえばステロイドや脾臓摘出などの通常の治療に反応しなかった特発性血小板減少性紫斑病患者の33~54%が,B細胞表面抗原のCD20に対するモノクローナル抗体(リツキシマブ)に反応する.
骨髄で生じる自己反応性B細胞,特に全身に普遍的に発現している細胞表面抗原に対する自己反応性B細胞は,クローン除去か受容体エディティングのどちらかの機序によって,骨髄中で除去される.種々のモデルマウスの研究で受容体エディティングが,骨髄での細胞表面自己抗原に対するトレランスのおもなメカニズムであることが明らかになりつつある.
(6)樹状細胞と自己免疫
抗原提示の中心である樹状細胞(dendritic cell: DC)が抗原を取り込むのは,ファゴサイトーシス,ピノサイトーシス,エンドサイトーシスなどによる.これらは抗原抗体複合体に対するFc受容体,糖蛋白に対するCLRs(c-type lectin receptors),微生物のPAMPs(pathogen-associated molecular patterns)を認識するTLRs(toll-like receptors)などを介して行われる.この際,DCは自己・非自己の認識をこれらの受容体を介して行っていることになる.すなわち定常状態でのDCは免疫学的には活性化されておらず,自己抗原に対する末梢の免疫寛容の一部は未熟DCによって維持されている.DC上のTLRが病原体の分子PAMPsを認識することによりDCは成熟するが,これはCLRsにより抑制されることもある.このCLRsを刺激するのは自己抗原または病原体の特異な糖鎖である.CLRsはDC,マクロファージ,内皮細胞などの抗原提示細胞に発現していて,エンドサイトーシスから抗原捕獲や抗原のクリアランスの役割を果たしている.代表的なCLRsにはDEC205(CD205)やDC-SIGN(CD209),Dectinなどがある.CLRsは糖鎖認識ドメインをもち,N結合型糖鎖やO結合型糖鎖を認識する.したがって,たとえば自己抗原の糖鎖修飾の変化などでCLRsへのシグナルが弱まることによりTLRsからのシグナルが強まり自己免疫応答が惹起される可能性もある.[山本一彦]
出典 内科学 第10版内科学 第10版について 情報
Sponsored by ![]()