
内科学 第10版 「冠血流量調節と心筋虚血」の解説
冠血流量調節と心筋虚血(虚血性心疾患)
a.冠循環の解剖
左右の冠動脈は,起始部では直径3〜5 mm程度であり,それぞれ心外膜側を房室間溝および心室間溝に沿って走行しながら,分岐を繰り返し直径0.5 mm以下の小動脈となってほぼ直角に心筋層内に入り,心筋内でもさらに分岐を繰り返して冠微小循環系を形成する.冠微小循環系は,直径100〜400 μmの小動脈,直径100 μm以下の細動脈を経て毛細血管へ移行する.毛細血管の長さは0.5〜1 mm,直径4〜5 μm程度であり,赤血球や白血球はその形態を変形させながら通過する.正常の心筋組織では,1 mm2あたり2000本以上の毛細血管が存在しており,そのうち約7割が機能している.1本の心筋線維に対し,おおよそ1本の毛細血管が存在し,動脈血液中の酸素濃度が低下するような状態では,普段機能していない毛細血管も機能するようになり虚血に対応する.その後,静脈系は合流を繰り返し,大部分は冠静脈洞を経て右房に還流する.
b.冠循環の特徴と調節機構
冠循環の特徴は第一に,心臓が収縮・拡張を繰り返すため,エネルギー消費の最も大きな臓器であり,大量の血液供給(安静時約1 mL/g/分)を必要とすることである.さらに血液中からの酸素の引き抜き(酸素摂取)率も約75% (ほかの臓器は約25%)と高いことが知られている.しかし,他臓器への分配血液量の低下を防ぐために,必要最小限に抑えられている (心拍出量の約5%)ため,冠血流供給が制限されると心筋は容易に虚血に陥る.
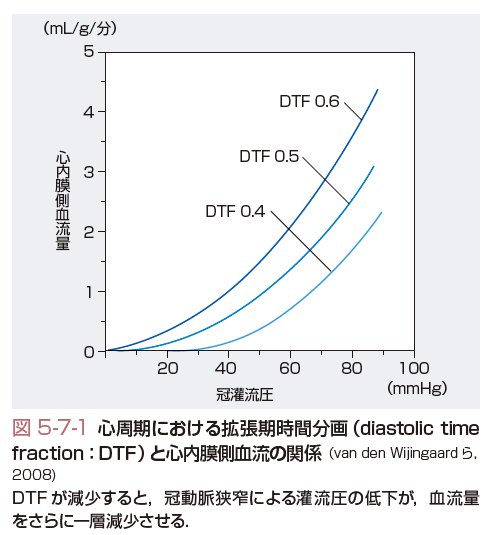
第二に,心臓では収縮期に心内膜側に高い圧力がかかるため,その部分の心筋酸素消費が大きいにもかかわらず,冠微小循環は収縮期に圧迫され,十分な組織灌流が得られない.つまり,冠微小循環では心周期のうち拡張期の限られた時間にしか血液が流れることができない.このような状況下において冠動脈狭窄などで冠血流量が低下すると,心内膜側・心外膜側血流比は1.1~1.4より容易に0.7くらいまで低下し,心内膜側より虚血が生ずる.さらに,心周期における拡張期時間分画 (diastolic time fraction:DTF)が減少すると,さらに冠血流量が低下するところとなる(図5-7-1) (van den Wijngaard,2008).
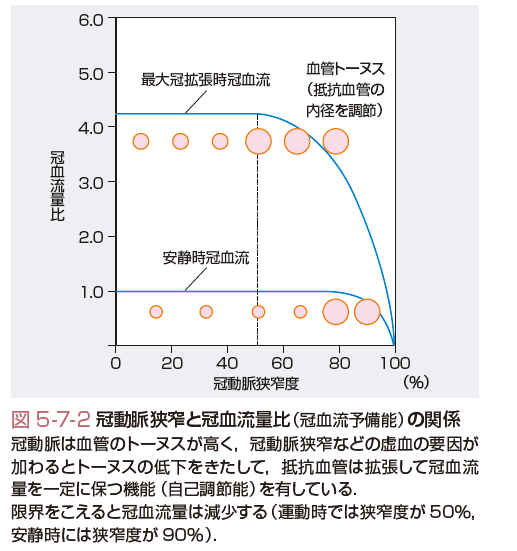
第三に,冠循環は脳循環とともに多少の灌流圧の変動にかかわらず冠血流量を維持する自己調節能(autoregulation) を有する点で,他臓器とは異なった循環調節機構を有する.すなわち,冠動脈における血管トーヌスが高く,虚血の要因が加わるとトーヌスの低下をきたし,冠血流量を一定に保つように調節している.心内膜下局所心筋短縮率または壁厚増加率が低下しはじめる臨界灌流圧(critical perfusion pressure)または自己調節能の限界は,40〜45 mmHgといわれている.安静時では,冠動脈の狭窄度が80%以上に進行すると冠血流量が低下しはじめる(図5-7-2).
心筋酸素消費量の増大に伴う冠血流量増加能力は,冠予備能(coronary flow reserve)とよばれている.冠動脈を数秒間閉塞した後,血流を再開させると最大の冠血流増加作用(反応性充血)が得られる.アデノシンやパパベリンによる冠血流増加作用は反応性充血に匹敵することから,臨床的にはこれらの薬剤を用いて,冠予備能の評価を行う.冠予備能は冠動脈の狭窄度が50%以上程度より低下がはじまる(図5-7-2).
冠循環は種々の特性を有しているが,その性質は抵抗血管と心筋細胞と酸素やグルコースなどの物質を直接交換する毛細血管に依存している.
c.冠血管トーヌス(coronary vascular tone)の制御
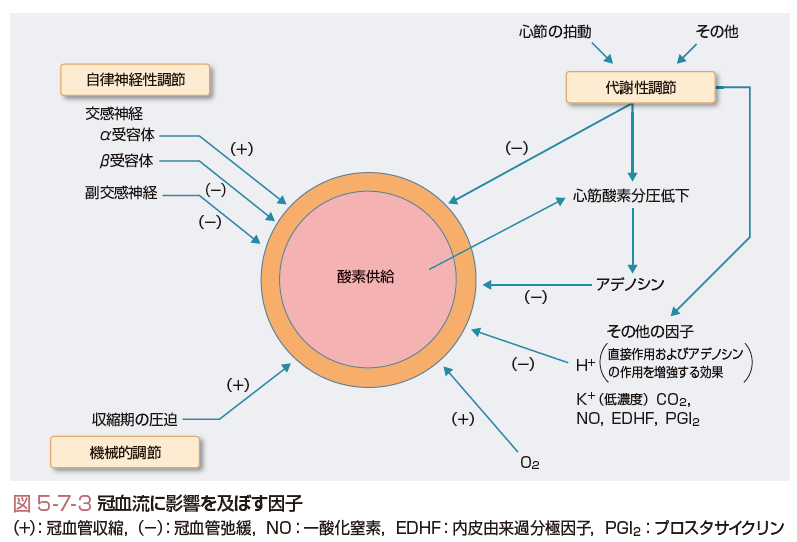
冠動脈において抵抗血管として機能するのは,直径100〜400 μmの小動脈と直径10〜100 μmの細動脈である.冠血管トーヌスは,おもに平滑筋自体の特性にて制御されているが,代謝性,内皮依存性,筋原性,神経・体液性因子の影響も受け,血管の太さによりおもに作用する機序が異なる.
i)冠血管トーヌスの代謝性調節
冠血流量は心筋酸素需要に応じて供給されるため,心筋の代謝を鋭敏に反映する物質が冠血管トーヌスを規定すると考えられている.心筋組織でのpH,PO2などは心筋の代謝を鋭敏に反映し,pH,PO2が低下すれば冠血流量が増加し,PCO2が上昇すれば冠血流量が 増加する.心筋虚血時に産生される水素イオン(H+)は直接冠血管を拡張させることで冠血流量を増加させる.また,カリウムイオン(K+)は冠血管を拡張させるが,高濃度では逆に冠血管を収縮させる.しかし実際には,K+はすぐに心筋細胞内に取り込まれるため冠血流量に大きく関与しているとは考えにくい.これらの因子による代謝性血管拡張は30 μm以下の細動脈で起こる.
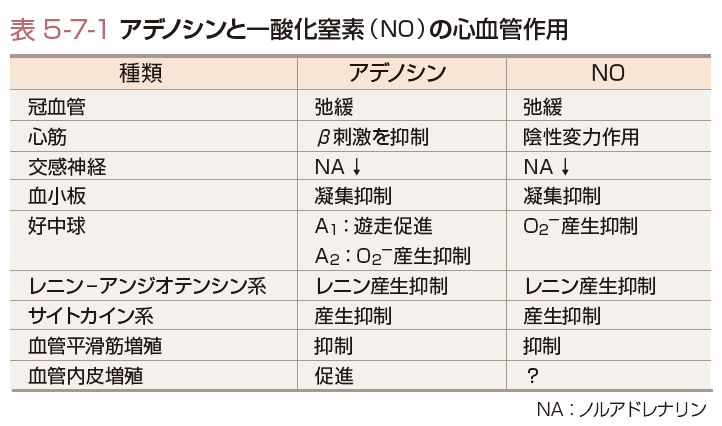
一方,ATPの代謝過程で生じるアデノシンは,心臓,腎臓,脳を含めた多くの器官において虚血や低酸素状態などの代謝性ストレス状態に陥ったときに産生が亢進し血管を拡張させ,組織の酸素需要-供給のバランス維持に働く.アデノシンはおもに直径50 μm以下の冠微小血管に作用して冠血管抵抗を低下させる.アデノシンは容易に細胞膜を通過し,細胞外に出てA1,A2A,A2B,A3の4種類の各受容体に結合し,多岐にわたる心血管保護作用を発揮し(表5-7-1),内因性の強力な心筋保護現象である虚血プレコンディショニング現象もアデノシンを介することが知られている.心筋虚血時には,心筋に存在するA1受容体刺激により,サイクリックAMP(cAMP)の合成が阻害され,心筋収縮を抑制し,冠動脈に存在するA2受容体刺激により,cAMPの合成が促進され,冠血管を拡張させる.さらに,アデノシンは血管内皮細胞から一酸化窒素(nitric oxide:NO)の分泌を促進し,ATP感受性K+チャネル(KATP)チャネルを介した血管拡張作用も有する(Hein ら,1999).アデノシンは,冠血管に対して耐性が生じにくく,冠血流量の体液・代謝性調節の主役の1つであると考えられている.
また,心筋虚血や低酸素により細胞内ATPレベルが低下するが,このATPレベル低下に呼応してKATPチャネルが開口し,冠血管抵抗が低下する.KATPチャネルが開口すると血管内皮内外での電気的・化学的勾配によりCa2+流入が増加し,内皮型NO合成酵素(eNOS)が活性化されてNO産生量を増加させることが知られている.KATPチャネルが作用する血管径はアデノシンと比較するとやや太く,100 μm程度の小動脈とそれ以下の細動脈であると考えられている.これらアデノシン,KATPチャネル開口およびNOの三者が協調的に冠血流量調節に働いている.
ⅱ)冠血管トーヌスの内皮依存性調節
NOは血流の存在する血管壁のshear stress(ずり応力)により内皮から恒常的に産生される.血流依存性の血管拡張反応はNOによるものと考えられており,直径100〜200 μmの比較的細い小動脈をおもに拡張させる.また,アセチルコリン,ヒスタミン,ブラジキニン,血小板 (セロトニン,ADP)などが血管内皮細胞の受容体に結合することにより,Ca2+の流入をもたらし,血管内皮細胞に局在するeNOSを活性化して,アルギニンからNOを継続的に産生する.産生されたNO は直ちに血管平滑筋に取り込まれ,可溶型グアニル酸シクラーゼの活性化によりサイクリックGMP(cGMP)が産生され,cGMP依存性蛋白質リン酸化酵素の活性化を介して血管平滑筋を弛緩させるなど,アデノシンと同様にさまざまな心血管保護作用を惹起する(表5-7-1).
また,内皮由来過分極因子 (endothelium-derived hyperpolarizing factor:EDHF)は生理活性物質による血管平滑筋の細胞膜のCa2+活性化型K+チャネル開口や,血管内皮細胞の膜電位変化がギャップ結合を介して平滑筋細胞に伝搬されることで血管を弛緩させると考えられている.EDHFの産生刺激因子はNO産生刺激因子の多くと共通しており,活性酸素(ROS)などによりNO産生が阻害されると代償性にその役割が大きくなる.EDHFの血管拡張作用はNOに比して,より直径が細い100 μm以下の抵抗血管において認められる.EDHF は不明の点が多いが,アラキドン酸のチトクロームP450依存性の代謝産物であるepoxide 11,12-epoxy-eicosatrienoic acid,K+イオン,H2O2などがその本体の1つではないかと考えられている.
そのほか,血管内皮のシクロオキシゲナーゼを介して産生されるプロスタサイクリン(PGI2)は血管平滑筋細胞のcAMPを介して,C型ナトリウム利尿ペプチドはcGMPを介してそれぞれ血管を弛緩させることでトーヌス調節に関与している.一方,血管内皮細胞はエンドセリンを産生放出して,血管平滑筋に作用し血管を収縮させる.このように冠血管内皮細胞は血流由来の諸因子のシグナルを感知するとともに,種々の血管作動物質を放出して冠血管のトーヌスを調節する能力を有する.
ⅲ)冠血管トーヌスの筋原性調節
筋原性反応は,内圧が高いと収縮し,内圧が低いと拡張する反応で,100 μm以下の冠細動脈で認められる.内皮を剥離しても生じることから,平滑筋によって惹起されると考えられている.
ⅳ)冠血管トーヌスの神経・体液性調節
冠動脈は全身の動脈のなかで最も豊富に神経が分布しており,交感神経系と副交感神経系の両者から支配を受けている.交感神経は無髄運動神経線維を有し,細動脈レベルに至るまで分布し,中膜平滑筋細胞に入り込む.近位部の冠血管ではα交感神経刺激による血管収縮作用が優位である.太い冠動脈ではα1受容体を介して血管が収縮し,抵抗血管ではα1およびα2受容体の双方を介して血管が収縮する.さらに遠位部の血管では,α作用が消失していく.α2受容体刺激は内皮由来のNO産生を惹起するが,両者の総和としては血管収縮作用が現れる.一方,β受容体は血管拡張性に作用し,近位の太い血管はβ1受容体が支配し,細い抵抗血管はβ2受容体が支配する.α交感神経受容体刺激による冠血管収縮作用の約70%は,太い冠動脈の収縮によるが,残りの30% は冠抵抗血管に起因することが示されており,冠抵抗血管はβ交感神経のみならず,α交感神経活性による制御も受けており,互いが拮抗して冠血流量を調節している.副交感神経は太い血管の外膜のみに分布し,運動性および知覚性の神経線維を含み,副交感神経を刺激すれば太い冠血管は拡張する.副交感神経刺激ではアセチルコリンの放出により,内皮由来NO産生を介して冠血管は拡張するが,中膜平滑筋の収縮作用も併せもつ.冠血管内皮機能が正常な場合,総和として冠血管拡張性に作用するが,動脈硬化などにより内皮機能が障害されている場合は冠血管収縮性に作用する.この作用は,冠攣縮性狭心症発症に深く関連している.
(2)冠血流低下が心機能に及ぼす影響
a.心筋のエネルギー代謝の変化
心臓はポンプ機能を維持するために,毎日約5 kgのアデノシン5′-三リン酸 (ATP)を産生している.正常心筋では,その大部分がミトコンドリアにおける酸化的リン酸化により産生されている.酸化的リン酸化の過程には常に大量の酸素が必要であり,冠血流に依存している.産生されたATPは心筋細胞内でクレアチンと結合し,クレアチンリン酸として細胞内に貯蔵されるが,この反応を触媒するのが細胞質内に存在するクレアチンキナーゼ(CK)である.心筋収縮やカルシウムハンドリングの維持,イオンの能動輸送にはATPが必要であるが,そのATPの供給源はクレアチンリン酸が細胞質内のCKにより還元されたものと考えられている.心筋は骨格筋に比し,クレアチンリン酸の貯蔵が少ないため,心筋虚血や心不全によって容易に枯渇してしまい,収縮障害や心筋壊死に陥る.心筋虚血状態では,ATP産生は脂肪酸のβ酸化から酸素を利用しない解糖系へシフトする.これは嫌気性解糖系であり,グルコースは嫌気性解糖を受けピルビン酸となり,産生されたピルビン酸はTCA回路に入らずにLDHの還元作用により乳酸に変換される.嫌気的条件下では乳酸は利用できないことから,心筋内に蓄積した乳酸は最終的には冠静脈血中に流出する.心筋虚血時においては,解糖によるATP産生量は少ないが,細胞膜機能の維持に重要な役割を果たしていると考えられる.心筋梗塞をきたすような高度虚血では,脂肪酸のβ酸化と解糖系はともに障害され,ATP産生が停止し,細胞内ATPの枯渇,水素イオンや乳酸の蓄積によりカルシウムハンドリング異常や細胞内pH異常により心筋細胞は壊死する.心筋虚血時には,まず心筋代謝異常が生じ,続いて拡張障害,収縮障害,電気生理学的障害,そして胸痛,心筋壊死が出現する(虚血カスケード).
b.収縮機能の変化
ⅰ)冠血流低下による心筋収縮の変化
冠動脈狭窄や血圧低下により冠灌流圧が40〜45 mmHg程度に低下し,冠血流量が自己調節能の範囲以下に減少すると特に心内膜側において,収縮期壁厚増加率や収縮期局所心筋長短縮率が低下する.この結果は,冠血流量が減少して自己調節能が限界に達した状態では,心筋へのエネルギー供給の指標である冠血流量と,心筋のエネルギー需要を反映する心筋収縮機能とが密接に関係していることを示唆するものであり, perfusion-contraction matching(灌流-収縮適合)とよばれる.
ⅱ)冠血流途絶による心筋収縮の変化
冠動脈を結紮すると灌流領域中心部の心筋短縮率が結紮後3~5心拍目から低下し,結紮10秒後には30%低下する.虚血がさらに持続すると心筋は収縮しなくなり,逆に収縮期を通じて伸展されるようになり(収縮期膨隆:systolic bulge),Frank-Starling機構が作動しなくなる.心筋長の変化に呼応して,虚血領域の拡張末期の左室壁厚(end-diastolic wall thickness:EDW)が減少し,収縮期の壁増厚(wall thickening)は低下する.結紮1分後には,EDW は結紮前よりも薄くなり(菲薄化),収縮期を通じて壁厚が増さなくなる.心筋虚血部位のこのような速やかな筋収縮障害は,無機リンの蓄積,アシドーシスの出現,Ca2+代謝異常,冠灌流圧の低下などに起因することが知られているが,これに加えて,局所の後負荷不整合がさらに心筋収縮を抑制する.
c.拡張機能の変化
心筋虚血時の左室拡張機能の変化として重要なものは,拡張早期にみられる弛緩速度の低下と,拡張中期から後期における拡張期スティフネスの変化の2点である.一般的に,心筋虚血が起こると収縮期よりも拡張期の指標の方が先に障害される.
ⅰ)弛緩速度の低下
細胞膜の脱分極によってCa2+がトロポニンと結合すると,収縮蛋白が活性化され,心筋は収縮し,収縮蛋白に結合したCa2+が筋小胞体に取り込まれると収縮蛋白の結合は不活性化され,心筋は弛緩する.心筋虚血が起こるとATPが減少するが,筋小胞体へのCa2+の取り込みにはATPを要するため,収縮蛋白の不活性化が遅延し,弛緩速度が低下する.
ⅱ)拡張期スティフネスの変化
拡張期における心筋あるいは心室の硬さはスティフネスとよばれる(コンプライアンスの逆数).低酸素血症による心臓全体の虚血では,冠灌流圧が一定であれば冠血流量が増加するため, coronary turgorの増加(garden hose effect)や間質浮腫により拡張期スティフネスが増加する.冠血流途絶が1時間以上持続すると心筋のATPが枯渇してアクチン-ミオシン結合が解離できなくなり,不可逆性の虚血性拘縮のため心筋スティフネスは著明に増加する.
(3)心筋壊死巣形成の寄与因子
a.虚血時間
心筋虚血による心筋壊死は心内膜側心筋より生じ,虚血時間が長くなるにつれて心外膜側心筋に伝播する(wave front現象).ヒトの心筋梗塞では,発症後3~6時間以内に貫壁性梗塞が完成することから,経皮的冠動脈インターベンション(percutaneous coronary intervention:PCI)による早期再灌流が重要となる.
b.側副血行路
側副血行路が発達しているイヌと側副血行路が乏しいブタを比較すると,心筋壊死形成に要する時間が異なることが知られており,側副血流量は梗塞サイズの規定因子となる.
c.虚血プレコンディショニング(ischemic preconditioning:IP)
IPは,先行する短時間心筋虚血により,その後生じる長時間虚血による心筋細胞傷害に対して保護的に作用する現象である.急性心筋梗塞患者において,先行する狭心症発作を有する症例の予後や心機能が比較的良好であることは,経験的に知られていた.1986年に急性心筋梗塞モデルで実験的に報告され(Murry,1986),その後心筋だけでなくほかの臓器でも同様の現象が報告され,IPは臓器保護の観点から普遍的な概念となった.IPには,心筋保護効果が1~2時間で消失するearly phase IPと心筋保護効果が24~48時間後に再び出現するlate phase IPとがある.early phase IPは蛋白リン酸化が関与するのに対して,late phase IPは蛋白合成を伴った現象であると考えられている.early phase IPの分子メカニズムとして,心筋間質で増加したアデノシン,ノルアドレナリン,ブラジキニン,オピオイド,アンジオテンシンⅡなどが,それぞれアデノシンA1/A3受容体,α1受容体,ブラジキニンB2受容体,δ-オピオイド受容体,アンジオテンシンⅡタイプ1(AT1)受容体のトリガー受容体を刺激し,G蛋白によるホスホリパーゼCおよびホスホリパーゼD活性化,さらにプロテインキナーゼC(PKC)活性化を経て,ミトコンドリアKATPチャネル開口やmitochondrial permeability transition pore(mPTP)開口抑制により心筋保護作用を惹起することが知られている.また,PKCは,ecto-5′-nucleotidase(アデノシン産生酵素)をリン酸化・活性化する.このアデノシン産生酵素活性化により,IP中のアデノシン産生が増強され,より効果的にIPの心保護作用の獲得が可能となる.その他,p38MAPKおよび熱ショック蛋白(HSP)27などが関与することが報告されている.一方,late phase IPの分子メカニズムについてはiNOSを介するNO産生増加-HSP72-MnSOD連関およびPKC-アデノシン産生酵素誘導-アデノシン連関の相加および相乗作用により惹起されるものと考えられている.
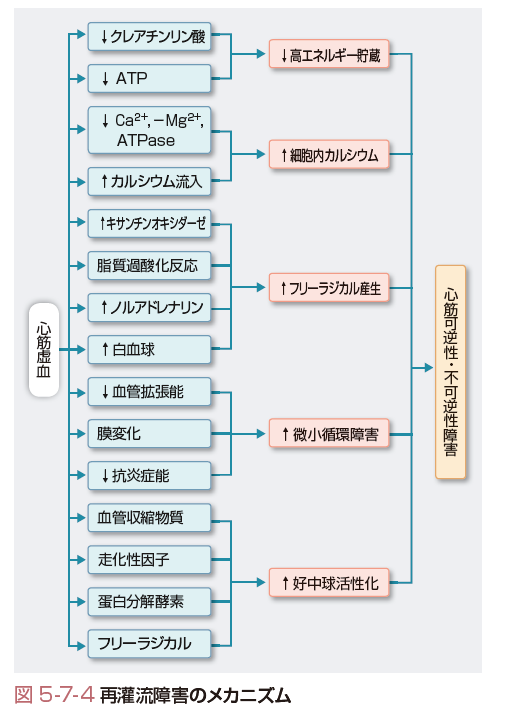
d. 再灌流障害
急性心筋梗塞発症早期の再灌流療法の普及により,心筋梗塞サイズが縮小することで死亡率が低下し,予後が大きく改善している.しかし,早期再灌流療法の施行にもかかわらず,心筋壊死,心筋スタニング,再灌流性不整脈,no reflow現象などさまざまな病態が生じることが明らかとなり,再灌流障害とよばれている.再灌流障害はCa2+過負荷,活性酸素・フリーラジカルの産生,血管内皮・好中球・血小板の活性化,エンドセリン,レニン-アンジオテンシン系,サイトカイン,交感神経活性化,小胞体ストレス,アポトーシス誘導などが複雑に関連して生じることが明らかにされている.
ⅰ)心筋スタニング
実験的に冠動脈を短時間結紮した後,再灌流すると組織学的にほとんど異常がないにもかかわらず心筋収縮能の低下が数時間~数日持続する.この再灌流後の心筋収縮不全は,1982年,Braunwald らによって“stunned myocardium(気絶心筋)”と命名された.臨床的には,虚血・再灌流後,気絶心筋と壊死心筋は混在するため,その回復過程には個体差がみられる.心筋スタニングのおもな原因として,心筋細胞内Ca2+過負荷とフリーラジカル発生が考えられている.
ⅱ)ハイバネーション
“hibernating myocardium(冬眠心筋)” 心筋ハイバネーションは,冠血流量が低下しても心筋は酸素供給に見合うように心機能を低下させ虚血性代謝変化を呈さない状態をいう.この概念は臨床的に,冠動脈狭窄のある患者が冠動脈バイパス術を受けた後,心機能の改善がみられることから提唱されたものである.一方,心筋ハイバネーションには繰り返す心筋虚血が関与しているとする考え方もある.Rahimtoolaらは,心筋ハイバネーションを「安静時には無症候であるが可逆的な心筋虚血が持続している左心室機能異常である」との仮説を唱えており,心筋ハイバネーションは種々の虚血状態よりなる病態である可能性がある.
ⅲ)no reflow 現象
冠動脈を結紮し,その後再灌流しても血流が回復しない領域が存在することが実験的に明らかになりno reflow現象として報告された.その後,臨床的にも心筋シンチグラフィや心筋コントラストエコー法をはじめとする画像診断の進歩により,急性心筋梗塞の多くの症例で生じており,心筋梗塞後の心機能や予後に影響を及ぼしていることが明らかにされている.no reflow 現象の原因は,血管内皮の腫脹,心筋細胞や間質の浮腫,Ca2+過負荷による心筋細胞の過収縮による毛細血管の圧迫,多核白血球や血小板による塞栓などが複雑に関与している.近年,PCIが盛んに施行されるようになり,プラークを機械的に刺激することにより,微小血栓,脂質成分,炎症細胞などが塞栓子となりno reflow 現象を呈する症例が存在するため,カテーテルおよび薬剤を用いた予防法や治療法が試みられている.
ⅳ)左室リモデリング
心筋梗塞後リモデリングは,急性心筋梗塞発症後,左室内腔がしばしば進行性に拡大する現象であり,左室内腔の拡大は,梗塞発症数日以内に起こる梗塞部伸展(infarct expansion)と,梗塞発症数週間~数カ月~数年に及ぶ非梗塞部の心筋肥大, 心拡大によるものがある.左室リモデリングは,機械的な伸展負荷と神経・体液性因子によって引き起こされる.左室リモデリングが起こりやすい因子として,左室の20%以上の広範囲心筋梗塞,曲率半径の大きい前壁心筋梗塞,梗塞責任血管の閉塞,および貫壁性梗塞などがある.左室リモデリングのメカニズムとして,①レニン-アンジオテンシン系,②交感神経系,③BNP・ANP,④マトリックス・メタロプロテアーゼ(MMP)などの関与が報告されている.これらの因子の中でも,レニン-アンジオテンシン系が最大の因子で,アンジオテンシン変換酵素阻害薬やβ遮断薬,心房性ナトリウム利尿ペプチドなどの薬剤により,左室リモデリングが抑制される.[浅沼博司・北風政史]
■文献
Hein TW, Kuo L: cAMP-independent dilation of coronary arterioles to adenosine : role of nitric oxide, G proteins, and K(ATP)channels. Circ Res, 85: 634-642, 1999.
Murry CE, Jennings RB, et al: Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation, 1986, 74: 1124-1136, 1986.
van den Wijngaard JP, Kolyva C, et al: Model prediction of subendocardial perfusion of the coronary circulation in the presence of an epicardial coronary artery stenosis. Med Biol Eng Comput, 46: 421-432, 2008.
出典 内科学 第10版内科学 第10版について 情報
Sponsored by ![]()