
内科学 第10版 「IgG4関連疾患」の解説
IgG4関連疾患(リウマチ性疾患)
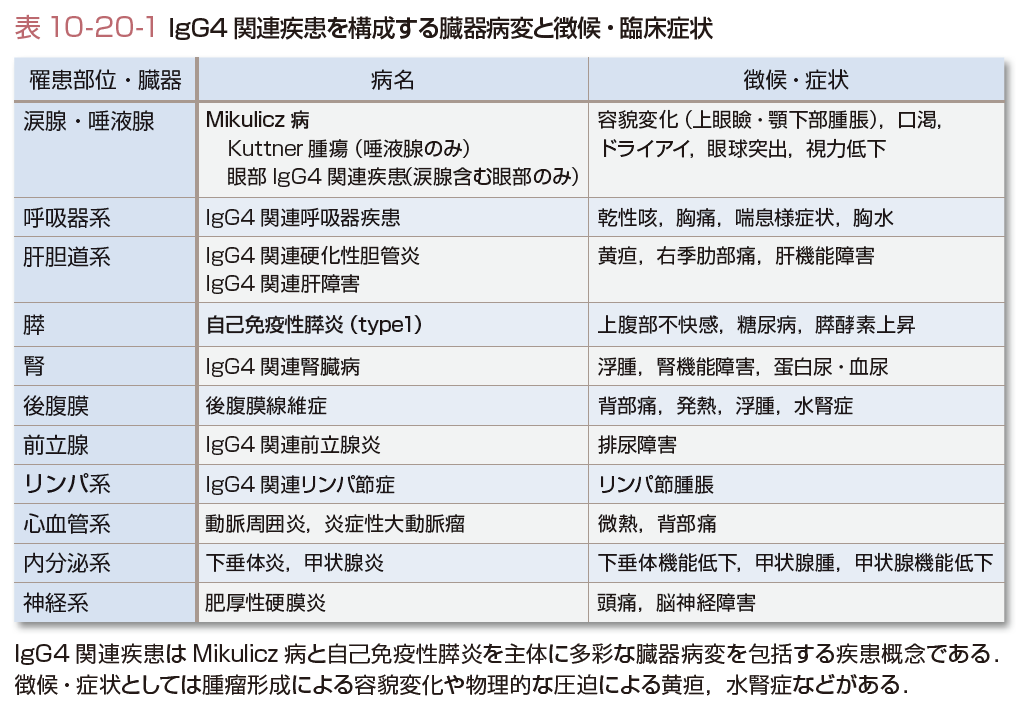
IgG4関連疾患は血清IgG4値の上昇と病変部へのIgG4陽性形質細胞の浸潤,線維化を主体とした腫瘤性・結節性・肥厚性病変を呈する慢性疾患である.IgG4-RDは自己免疫性膵炎(autoimmune pancreatitis:AIP)とMikulicz病(Mikulicz’s disease:MD)を2大病変とし,基盤に共通した病因・病態が想定されるわが国発の新たな疾患概念である(表10-20-1).グルココルチコイドへの良好な反応性や病変形成の時間的・空間的多発性も特徴である(Takahashiら,2010).
分類
IgG4-RDはMD,AIP,IgG4関連腎臓病・後腹膜線維症などの病態を,その病理組織学的共通性や重複罹患することから,全身性疾患として包括した概念である.したがって,単一臓器罹患の場合,臓器ごとの病名で呼称されることが多いが,IgG4-RDの各表現型としてとらえることもできる(表10-20-1).
原因・病因
病因は不明である.高ガンマグロブリン血症や非特異的な自己抗体の存在,グルココルチコイドが奏効することから自己免疫機序の関与が推測されているが,自己抗原・自己抗体の直接的な証明はなされていない.
疫学
全国調査の結果,患者数はAIPで約3000人(2007年),MDで約1100人(2010年)と報告されたが,今後の増加が予測される.IgG4-RDの発症年齢は50歳から60歳にピークがある.男女比はMDを除くと明らかに男性に多いが,MDでは性差は認められない.
病理(図10-20-1)
IgG4-RDに共通した病理所見はポリクローナルなリンパ球・形質細胞の著明な浸潤と線維化である.浸潤した形質細胞の多くはIgG4陽性である.胚中心の発達したリンパ濾胞の形成がみられ,好酸球浸潤が目立つ症例もある.AIPなどでの閉塞性静脈炎の存在も特徴的である.一方,好中球浸潤や肉芽腫病変は認められない.
病態生理
IgG4自体は補体活性化能・Fcγ受容体結合能を欠き,病変形成へのIgG4の直接的な関与は否定的である.しかし,高IgG4血症を生じるようにB細胞でのクラススイッチがIgG4優位に誘導されていることから,病変部でのTh2サイトカインへの偏移,あるいはIL-10の増加が推測される.事実,IgG4-RDの病変局所においてはTh2優位の免疫応答とIL-10産生にかかわる制御性T細胞の多数の浸潤が報告されている.また,制御性T細胞由来のTGFβが線維化を惹起している可能性も示唆される.
臨床症状
発熱,全身倦怠感などの全身症状はまれで,侵される罹患臓器由来の局所症状が主体である(表10-20-1)が,無症候性のことも少なくない.MDでの容貌変化,後腹膜線維症での水腎症,硬化性胆管炎での黄疸など腫瘤形成による圧迫などの物理的な症状・症候が先行した場合はこれらが診断のきっかけになる.しかし,病変が持続すると,MDでの口渇などの乾燥症状や間質性腎炎による腎機能障害,AIPでの膵内分泌機能低下による耐糖能障害が生じうる.
検査成績
1)臨床検査:
高ガンマグロブリン血症,好酸球増加,血清IgE上昇が認められる.ときに抗核抗体・リウマトイド因子が陽性であるが,疾患特異的な自己抗体は陰性である.血清IgG4高値はほぼ必発であり,135 mg/dL以上をカットオフとしてスクリーニングを行う(健常人40〜80 mg/dL)が,MDでは500 mg/dL以上もまれではない(Yamamotoら,2006).罹患臓器数の多い症例,特に腎病変合併例において低補体血症や免疫複合体上昇を認めることが多い.
2)画像(図10-20-2):
造影CTやMRIにより,罹患臓器のびまん性・限局性の腫大が検出される.特にIgG4関連腎臓病の診断には造影CTが有用である.またIgG4-RD診断時には必ず他臓器の罹患の有無をチェックする必要があり,全身のスクリーニングにはGaシンチグラフィやFDG-PET(18F-fluorodeoxyglucose positron emission tomography)検査の施行が勧められる.
診断
現在,IgG4-RDの包括的な診断基準が厚生労働省研究班中心に策定中であるが,要旨は①単一または複数の臓器の腫瘤性・結節性・肥厚性病変,②高IgG4血症(135 mg/dL以上),③リンパ球・形質細胞浸潤と線維化,およびIgG4/IgG陽性細胞比40%以上,あるいは10個以上/強拡大視野,の3条件を満たすことである.また,AIP・MDに関しては,それぞれ厚生労働省研究班・日本膵臓学会(2006年),日本シェーグレン症候群学会による診断基準が提唱されている(高橋ら,2010).
鑑別診断
IgG4-RDは腫瘤形成性の病変を呈することから,鑑別診断として悪性腫瘍やリンパ増殖性疾患,肉芽腫性疾患(サルコイドーシスなど)があげられる.可能な限り,病理組織学的な検索を行うことが勧められ,特にAIPでは膵癌,MDでは悪性リンパ腫・Sjögren症候群との鑑別が不可欠である.診断マーカーとしての血清IgG4値の有用性は広く認められているが,血清IgG4やIgG4陽性細胞数の増加は必ずしもIgG4-RDに特異的ではないことから,診断時には総合的に判断する必要がある.
経過・予後
MDやAIPとして診断され,その全身検索中,あるいはフォロー中に腎病変などの他臓器病変が発見されることが多い.MDの約半数では診断時に他臓器病変の合併(後腹膜線維症20%,膵 19%,腎 16%など)がみられる.グルココルチコイドへの反応性が良好であり,臓器・生命予後は一般に良好とされているが,新たな疾患概念であり,今後の慎重な経過観察を要する.
治療
第一選択はグルココルチコイドである.黄疸,水腎症などの進行性の臓器障害を呈している場合や,罹患臓器が複数の場合は絶対的な治療適応である.自覚症状を有する場合も治療適応であるが,無症候例では経過観察を行うこともある.単一臓器罹患の場合,グルココルチコイドの投与はプレドニゾロン換算で0.6 mg/kg/日から開始する.複数の臓器障害例では1 mg/kgまでの増量も考慮し,プレドニゾロンの初期投与量を2〜4週間継続後,2週間ごとに5 mgずつ減量していく.維持療法の要否に関しては結論が出ていないが,減量中止後の再燃がまれではないことから,5〜10 mg/日程度の継続投与が勧められる.グルココルチコイドの一次無効例はまれであるが,最近,減量困難例や再燃例が問題となっている.免疫抑制薬や抗CD20抗体などによる治療が試みられているが,有効性の評価は定まっていない.[髙橋裕樹]
■文献
高橋裕樹,山本元久:IgG4関連疾患.GUIDELINE膠原病・リウマチ−治療ガイドラインをどう読むか−(小池隆夫・住田孝之編),pp43-49,診断と治療社,東京,2010.
Takahashi H, Yamamoto M, et al: The birthday of a new syndrome: IgG4-related diseases constitute a clinical entity. Autoimmun Rev, 9: 591-594, 2010.
Yamamoto M, Takahashi H, et al: A new conceptualization for Mikulicz’s disease as an IgG4-related plasmacytic disease. Mod Rheumatol, 16: 335-340, 2006.
IgG4関連疾患(その他のリンパ増殖性疾患)
高IgG4血症と種々の組織でのIgG4産生形質細胞の増加による腫瘤形成や臓器障害を特徴とする原因不明の疾患である.[塚崎邦弘]
■文献
Dispenzieri A, Pittaluga S, et al: Diagnosis and management of disorders that can mimic lymphoma. In: Non-Hodgkin Lymphoma, 2nd ed (Armitage JO, Mauch PM, et al eds), pp 557-585, Lippincott Williams & wilkins, Philadelphia, 2009.
正木康史,梅原久範:IgG4関連疾患-新たな疾患概念-.臨床血液,52: 315-321,2011.
出典 内科学 第10版内科学 第10版について 情報
Sponserd by ![]()