
内科学 第10版 「多発性内分泌腫瘍症1型」の解説
多発性内分泌腫瘍症1型(MEN1型)(多発性内分泌腫瘍症)
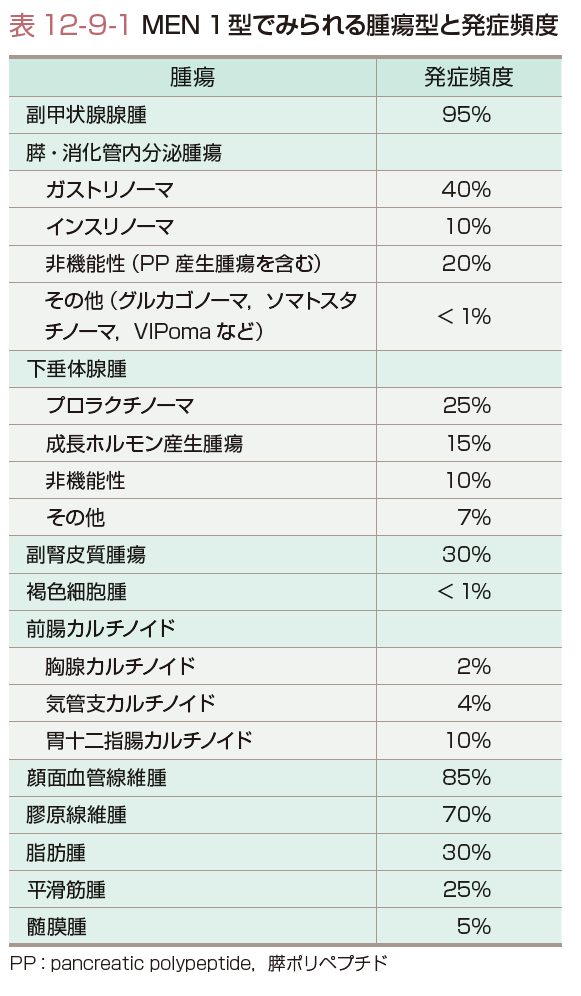
MEN 1型は同一家系内または同一人に発症して,下垂体,副甲状腺,膵Langerhans島の3つの内分泌腺に高い割合で腫瘍(または過形成)が多発する遺伝性疾患であり,常染色体優性遺伝形式をとる(表12-9-1).1954年Wermerらによって最初に記載されたことからWermer症候群ともよばれる.それ以外にもしばしば前腸由来カルチノイド腫瘍(神経内分泌腫瘍),脂肪腫,血管線維腫,副腎腫瘍なども合併する.本症の発生頻度は,人口3万人に1人と推定される.
病因
MEN 1型の原因遺伝子は第11染色体長腕(11q13)に局在し,1997年に分子クローニングによりMEN 1遺伝子が同定された.MEN 1遺伝子は10個のエクソンから構成され,このうちエクソン(2~10)が610個のアミノ酸からなるメニンをコードする.メニンは核蛋白質であり,転写因子Jun Dと結合してその機能を抑制する.MEN 1は胎生期では広範な組織で豊富に発現しているが,成体では脳を除いて低下する.
MEN 1は腫瘍の発生を抑える癌抑制遺伝子とされ,MEN 1型ではMEN 1遺伝子の変異は機能喪失型(“loss-of-function”)と考えられている.MEN 1型での腫瘍の発生機序として“two-hit”モデルが想定されている.すなわち家族性MEN 1型の家系および散発性MEN 1型のいずれでも一対のMEN 1遺伝子の片方に胚細胞変異が存在して(first hit),その機能が低下しているところへ,内分泌腺におけるもう一方の正常MEN 1遺伝子が体細胞変異を起こして(second hit),機能を喪失する結果,腫瘍が発生するというものである.MEN 1型での胚細胞変異はさまざまで,遺伝子全体に分布しており,“hot spot”とよべる部位がないのに対して,腫瘍で認められるMEN 1遺伝子の体細胞変異は正常MEN 1遺伝子を含む大領域欠失やヘテロ接合性の消失(LOH)であることが多い.しかし,なぜ特定の内分泌腺に限定して腫瘍化するのかの機序は不明である.
病態生理
1)副甲状腺機能亢進症:
副甲状腺では多発性に腺腫または過形成が高頻度(90~100%)に発生し,原発性副甲状腺機能亢進症の1~3%はMEN1型による.MEN 1型では副甲状腺機能亢進症が初発病変のことが多く,発症年齢は20歳代前半で性差を示さない.しかし無症候性高カルシウム血症のことが多いため,先に下垂体や膵腫瘍が発見されることが多い.高カルシウム血症が進行すれば汎発性線維性骨炎,再発性腎結石,消化性潰瘍,膵炎などを呈する.本症の副甲状腺病変は多腺性であり,複数の腺に同時か異時性に腺腫(過形成)が生じるが,癌への変化はまれである.血中PTHの増加と画像検査により副甲状腺病変の局在を証明する.
2)下垂体腫瘍:
下垂体では機能性(ホルモン分泌能をもつ)や非機能性腫瘍が発症し,その頻度は約40%である.下垂体腫瘍のうちMEN 1型によるものは5%以下である.機能性腫瘍にはプロラクチノーマが最も多く(60%),ついでGH産生腺腫(15%),ACTH産生腺腫(5%)がある.プロラクチノーマでは月経不順・無月経,乳汁漏出,GH産生腺腫では先端巨大症(手足の容積の増大,眉弓部の膨隆,下顎突出など),ACTH産生腺腫ではCushing症候群(満月様顔貌,中心性肥満,赤色皮膚線条など)の症状を呈する.それぞれ血中でプロラクチン,GHとソマトメジンC(IGF-Ⅰ),ACTHとコルチゾールの増加を示す.
非機能性腫瘍(25%)は,嫌色素性腺腫で下垂体機能低下症(やせ,食欲不振,低血糖,性欲低下,耐寒性低下など),視力障害,頭痛などの局所症状を呈する.単独あるいは複数の下垂体ホルモンの低下と頭部CTやMRIで下垂体腫瘍の存在を証明する.
3)膵内分泌腫瘍:
膵腫瘍の発症頻度は60%以上と副甲状腺機能亢進症についで高い.事実,40歳以上のMEN 1型の手術や剖検症例のほぼ100%で膵腫瘍が認められる.膵腫瘍は多発性で,機能性・非機能性あるいは良性・悪性が混在する.膵内分泌腫瘍で最も多いのはガストリン産生腫瘍(ガストリノーマ)で,ついでインスリン産生腫瘍(インスリノーマ)である.
ガストリノーマによるZollinger-Ellison症候群(繰り返す多発性の難治性消化性潰瘍),インスリノーマでは低血糖,その他の消化管ホルモン産生腫瘍としてVIP産生腫瘍(VIPoma)ではWDHA症候群(水様性下痢,低カリウム血症,無酸症),グルカゴノーマでは耐糖能異常や融解性移動性紅斑を呈する.それぞれ,血中のガストリン,インスリン,VIP,グルカゴンの高値と画像検査で腫瘍の存在を証明する.その他pancreatic polypeptide(PP),ソマトスタチン,カルシトニンなどを産生するが,無症状である.セロトニン産生神経内分泌腫瘍(カルチノイド腫瘍)ではセロトニンを代表とする種々の生理活性物質を産生・分泌してカルチノイド症候群を呈することがある.まれにACTHやGRHを産生してそれぞれCushing症候群や先端巨大症を呈する異所性ホルモン症候群を合併することもある.
診断
MEN 1型における内分泌腫瘍の診断は,散発性の症例と同じである.若年(10歳代)発症の原発性副甲状腺機能亢進症をみた場合,MEN 1型の可能性を考え,家族歴の聴取や下垂体や膵の内分泌腫瘍を念頭においてスクリーニング試験を行う.ガストリノーマではセクレチンやカルシウム刺激試験による血中ガストリンの過剰反応がみられる.インスリノーマでは72時間絶食試験による低血糖誘発時の血中インスリンの抑制がみられない.また,インスリノーマやガストリノーマの術前局在診断には選択的動脈内刺激薬注入試験(SASI)が行われる.栄養血管刺激後の肝静脈血サンプリングでそれぞれインスリンやガストリンの過剰反応がみられる.MEN 1型が疑われた場合,各内分泌病変に応じた生化学検査,内分泌検査,画像検査を定期的に行うが,最終的にはMEN 1遺伝子解析が必要となる.
MEN 1遺伝子の遺伝子診断はMEN 1型を疑診する患者の補助診断やMEN 1型家族での発症前の保因者診断として有用である.通常,家族性(80~90%),散発性(約70%)に変異が同定される.
MEN 1遺伝子変異をもつ人の生涯発症率(浸透率)はほぼ100%と考えられる.したがって,本症診断のための遺伝子検査は有用であり,患者自身だけでなく,発症の可能性がある家族での疾患の早期発見,早期治療に役立つ.しかし,遺伝子検査の施行にあたっては患者と家族に対する十分な説明と同意が必要である.
治療・予後
MEN 1型の下垂体腫瘍は,プロラクチノーマのドパミン作動薬による薬物療法を除いて経蝶形骨洞手術の適応である.副甲状腺腺腫は多発性であるため,腺腫摘出後も再発しやすい.したがって,手術時には3腺全摘と1腺半分切除する亜全摘術か,4腺摘出と一部自家移植の方法をとる.術中の超音波検査や迅速PTH測定は副甲状腺病変の局在や摘出の成否の判定に有用である.膵病変も多発性で腺腫や癌が混在するため,手術に際して,術中超音波や術者による触診によって腫瘍の局在を確認し,核出術や膵の部分切除を行う.
各腫瘍の多くは良性であるため予後は良好である.対症療法として,ガストリノーマに対してはH2受容体拮抗薬やプロトンポンプ阻害薬が,またインスリノーマに対してはジアゾキサイドやオクトレオチドが有効である.しかし,膵・消化管や胸腺・気管支の神経内分泌腫瘍(カルチノイド腫瘍)を合併した場合,悪性で転移することがあるため,予後は不良となる.[平田結喜緒]
■文献
Brandi ML, Gargel RF, et al: Guideline for diagnosis and therapy of MEN type1 and type2. J Clin Endocrinol Metab, 86: 5658-5671, 2001.
Gargel RF, Marx SJ: Multiple endocrine neoplasia. In: Williams Textbook of Endocrinology(Larsen PR, Kronenberg HM, et al eds),pp1717-1762, WB Saunders, Philadelphia, 2008.
Hoff AO, Gargel RF: Multiple endocrine neoplasia type2. In: Endocrinology(DeGroot LJ, Jameson JL eds),pp3533-3550, Elsevier Saunders, Philadelphia, 2005.
Thakker RN: Multiple endocrine neoplasia type1. In: Endocrinology(DeGroot LJ, Jameson JL eds),pp3509-3531, Elsevier Saunders, Philadelphia, 2005.
出典 内科学 第10版内科学 第10版について 情報
Sponsored by ![]()