
内科学 第10版 「体質性黄疸」の解説
体質性黄疸(肝・胆道の疾患)
体質性黄疸は肝細胞の先天性ビリルビン代謝異常により発症する黄疸である.ビリルビンおよび代謝機構がビリルビンと共通する有機陰イオンの代謝異常をみるが,日常臨床で用いられるビリルビン以外の肝機能検査には異常を認めない.高非抱合型ビリルビン(間接ビリルビン)血症をきたすものと高抱合型ビリルビン(直接ビリルビン)血症をきたすものに分類される.
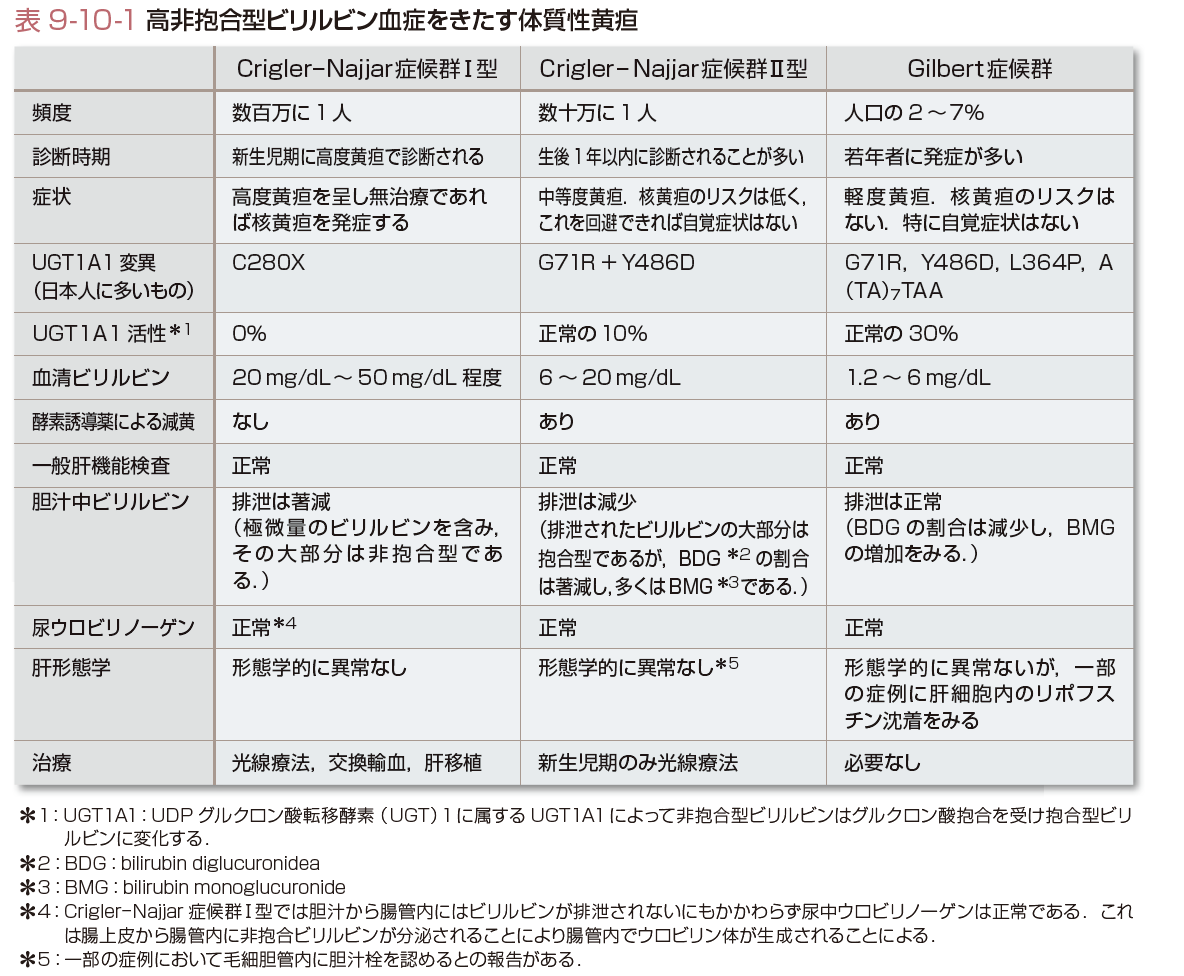
(1)高非抱合型ビリルビン血症をきたす体質性黄疸(表9-10-1)
分類
高非抱合型ビリルビン血症をきたす体質性黄疸は黄疸の程度から,Crigler-Najjar症候群Ⅰ型(ビリルビン濃度が20 mg/dL以上50 mg/dL程度まで),同症候群Ⅱ型(ビリルビン濃度が6 mg/dL以上20 mg/dL未満)とGilbert症候群(ビリルビン濃度が1.2 mg/dL以上6 mg/dL未満)に分類される.
疫学
Crigler-Najjar症候群Ⅰ型はきわめてまれである(出生100~数百万人に1人程度との記載もあるが正確な頻度は不明).わが国では数家系が報告されている.同症候群Ⅱ型の発生頻度は数十万人に1人とされている.Gilbert症候群はまれではなく人口の 2~7%にみられる.
病理
Crigler-Najjar症候群Ⅰ型,Ⅱ型の肝臓は形態学的に正常であるが,Ⅱ型では組織学的に毛細胆管内に胆汁栓を認めることもある.Gilbert症候群も形態学的に正常であるが,一部の症例に肝細胞内のリポフスチン沈着をみる.
病因・病態生理
Crigler-Najjar症候群Ⅰ型,Ⅱ型とGilbert症候群はビリルビンのグルクロン酸抱合酵素活性の低下あるいは欠損により黄疸を呈する.グルクロン酸抱合酵素であるUDPグルクロン酸転移酵素群(UGTs)にはUGT1とUGT2の2つのファミリーがあり,UGT1に属するUGT1A1によって非抱合型ビリルビンはグルクロン酸抱合を受け抱合型ビリルビンとなる.UGT1A1遺伝子のさまざまな変異/多型は種々の程度のUGT活性を作り出す.
Crigler-Najjar症候群Ⅰ型は先天的にUGT1A1活性が欠如している.UGT1A1遺伝子変異としてわが国では840C>A:C280Xのホモ接合体の報告が多い.同症候群Ⅱ型のUGT1A1活性は正常の約10%である.UGT1A1遺伝子に多くの変異が報告されているが,わが国では211G>A:G71Rと1456G>A:Y486Dの二重ミスセンス変異が多い.Gilbert症候群はUGT1A1活性が正常の約30%である.Gilbert症候群はUGT1A1遺伝子のコード領域のミスセンス変異(211G>A:G71Rが多い)とプロモーター領域のTATA boxのA(TA)6TAAがホモ接合体のA(TA)7TAAになることが原因とされている(Takeuchiら, 2004).コード領域の変異は日本人を含む東アジアからの報告が多く,欧米人ではまれとされている.
臨床症状・検査成績
Crigler-Najjar症候群I型は出生直後から高度黄疸(血清ビリルビン濃度は20 mg/dL以上50 mg/dL程度)を呈し,放置すれば核黄疸を発症し予後は不良である.胆汁には微量のビリルビンが排泄されるのみで便中ウロビリン体は著減するが,尿中ウロビリノーゲンは正常である.同症候群Ⅱ型は新生児期以降の黄疸持続で発症するが,幼児期の発症もある.乳児期までは核黄疸のリスクがあるが光線療法などで回避すれば正常に成長する.血清ビリルビンは6 mg/dL以上20 mg/dL未満であるが空腹で40 mg/dL程度まで上昇することがあるためI型との鑑別を要することがある.フェノバルビタールなどの酵素誘導薬は減黄に効果があり,Ⅰ型との鑑別に有用である.Gilbert症候群の黄疸は思春期以降に気付かれることが多い.多くは無症状だが軽度の倦怠感や右季肋部痛を訴えることもある.血清ビリルビン濃度は1.2 mg/dL以上6 mg/dL未満である.ビリルビン濃度は変動が大きく,基準値の範囲内に入ることもある一方,空腹時にはビリルビン濃度が上昇する.48時間低カロリー試験(400 kcal/日)によりビリルビン濃度が2倍程度の上昇をみることは診断の一助となる.これらの症候群ではほかの一般肝機能検査は正常である.
診断・鑑別診断
成人の高間接ビリルビン血症をきたす病態には,溶血性黄疸,シャント高ビリルビン血症,Crigler-Najjar症候群Ⅱ型,Gilbert症候群がある.Crigler-Najjar症候群Ⅱ型とGilbert症候群は溶血性黄疸やシャント高ビリルビン血症を否定すればビリルビン濃度の差で診断する.溶血性黄疸は貧血,LDH上昇,ハプトグロビン低下などを伴うことにより鑑別される.シャント高ビリルビン血症は無効造血に由来するビリルビンが増加した病態で,その原因が不明の原発性と悪性貧血やサラセミアなどが原因の二次性がある.無効造血の原因となる疾患の診断に加え,鉄動態検査結果から診断される.新生児から乳児期のすべて黄疸はCrigler-Najjar症候群Ⅰ型,Ⅱ型と鑑別対象となる.
経過・予後・治療
Crigler-Najjar症候群Ⅰ型では核黄疸予防に光線療法が有効である.継続して光線療法を行うことにより生命予後を改善するが思春期以降は皮膚の肥厚により効果が減少する.ほかの減黄法として交換輸血,ビリルビン吸着がある.酵素誘導薬は効果がない.肝移植により根治が可能である.同症候群Ⅱ型に対しては新生児期の光線療法は有効で,成長すると核黄疸のリスクはほぼなくなる.酵素誘導薬の効果があり,美容上の問題があるときには考慮する.Gilbert症候群は治療不要である.
UGT1A1はビリルビン代謝以外にも薬物,生体内物質,発癌性物質の代謝にも関与している.抗癌薬である塩酸イリノテカンはUGT1A1を含むUGTファミリーにより代謝されるが,Gilbert症候群でみられるA(TA)7TAAやG71Rでは血中塩酸イリノテカン濃度が高くなり副作用が出やすいため投与前にUGT1A1遺伝子多型の確認が推奨されている.またGilbert症候群はUGT1A6,1A7, 1A9の遺伝子多型ともリンクし,これらの遺伝子多型も薬物代謝遅延や副作用増強を引き起こす(Lankischら, 2008).UGT1A1変異がさまざまな発癌物質代謝が遅延するのでGilbert症候群におけるUGT1A1の遺伝子多型と発癌リスクの検討が必要である.
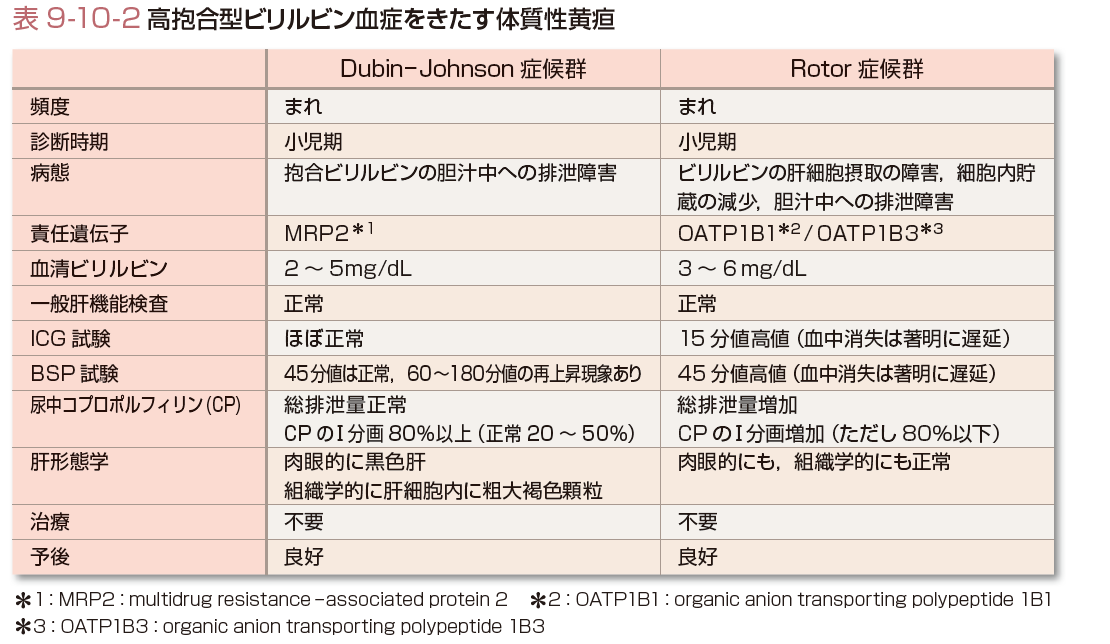
(2)高抱合型ビリルビン血症をきたす体質性黄疸(表9-10-2)
分類
高抱合型ビリルビン血症をきたす体質性黄疸にはDubin-Johnson症候群とRotor症候群がある.
疫学
Dubin-Johnson症候群とRotor症候群はともにまれであり,正確な発生頻度は不明である.わが国ではDubin-Johnson症候群は文献的に約400例程度の報告があり,Rotor症候群は約百数十例程度の報告がある.
病理
Dubin-Johnson症候群は肉眼的に黒色肝を呈する.組織学的に肝細胞内に粗大褐色顆粒を認め,この顆粒は肝炎時に消失する.Rotor症候群の肝臓は形態学的異常を認めない.
病因・病態生理
高抱合型ビリルビン血症をきたす体質性黄疸はビリルビン抱合後のビリルビン代謝過程の障害で発症する.抱合型ビリルビンは毛細胆管側肝細胞膜に運ばれ,膜上の輸送蛋白であるmultidrug resistance-associated protein 2 (MRP2)により胆汁中に排泄される.Dubin-Johnson症候群はこのMRP2が欠損しており(Kartenbeck ら, 1996),胆汁中への抱合ビリルビンの排泄障害のため黄疸を呈する.胆汁酸の排泄は正常であることから胆汁うっ滞と鑑別される.Rotor症候群はビリルビンの肝細胞摂取,細胞内貯蔵,胆汁排泄のいずれもが障害されている.肝細胞類洞側細胞膜上に存在するorganic anion transporting polypeptide1B1(OATP1B1)およびOATP1B3をコードする遺伝子の変異がRotor症候群の原因であるとの報告がある.
臨床症状・検査成績
Dubin-Johnson症候群,Rotor症候群ともに黄疸以外の症状がなく,小児期から成人後に軽度の黄疸(血清ビリルビン濃度は2〜5 mg/dL程度,Rotor症候群がやや高いという報告もある)で気付かれる.ビリルビン濃度以外の一般肝機能検査は正常である.これらの鑑別には色素排泄試験であるインドシアニングリーン(indocyanine green:ICG)試験やブロモスルホフタレイン(bromosulphophthalein:BSP)試験と尿中コプロポルフィリン(CP)排泄の解析が有用である.Dubin-Johnson症候群ではICG試験はほぼ正常(ないし軽度上昇)である.BSP試験では45分値はおおむね正常であるが60~180分値では特徴的な再上昇現象を認める.BSP再上昇はMRP2の機能異常とその代償機構によると考えられるグルタチオン抱合型BSPの血中への逆流を反映している.一方,Rotor症候群ではICG試験,BSP試験ともに高度の排泄遅延を認め,BSP再上昇現象は認めない.尿中CPの排泄に関してはDubin-Johnson症候群ではCP総排泄量は正常でCPのⅠ分画が80%以上(健常人のCPのI分画は20〜50%程度)である.Rotor症候群では尿中CP総排泄量は著増し,Ⅰ分画の割合は増加するが80%以下にとどまる.
診断・鑑別診断
高抱合型ビリルビン(直接ビリルビン)血症を示すが,ほかの一般肝機能検査や画像検査が正常であれば,体質性黄疸であるDubin-Johnson症候群,Rotor症候群の可能性が高いのでICG試験,腹腔鏡,尿中CPの排泄を調べる.
経過・予後・治療
Dubin-Johnson症候群,Rotor症候群は予後良好で治療不要であるが,有機アニオン系薬物の胆汁排泄遅延を起こしている可能性を想定すべきである.[上硲俊法]
■文献
Takeuchi K, Kobayashi Y, et al: Genetic polymorphisms of bilirubin uridine diphosphate-glucuronosyltransferase gene in Japanese patients with Crigler-Najjar syndrome or Gilbert's syndrome as well as in healthy Japanese subjects. J Gastroenterol Hepatol, 19: 1023-1028, 2004.
Lankisch TO, Schulz C, et al: Gilbert's Syndrome and irinotecan toxicity: combination with UDP-glucuronosyltransferase 1A7 variants increases risk. Cancer Epidemiol Biomarkers Prev, 17: 695-701, 2008.
Kartenbeck J, Leuschner U, et al: Absence of the canalicular isoform of the MRP gene-encoded conjugate export pump from the hepatocytes in Dubin-Johnson syndrome. Hepatology, 23: 1061-1066, 1996.
出典 内科学 第10版内科学 第10版について 情報
Sponserd by ![]()