
内科学 第10版 「心血管リモデリング」の解説
心血管リモデリング(循環器疾患の主要病態)
(1)心室リモデリング(ventricular remodeling)
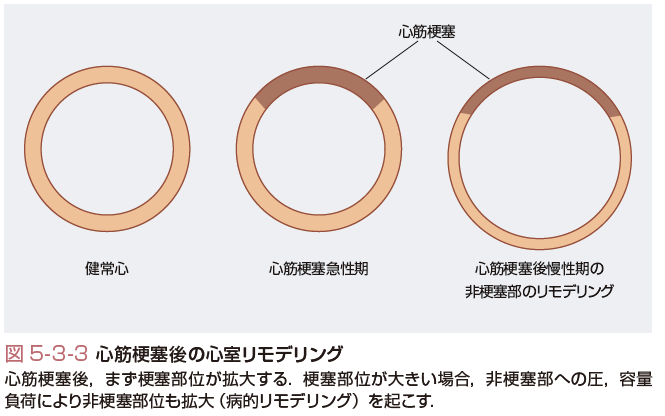
過剰な負荷,または心筋収縮力が低下した場合,心臓はその拍出量を維持するためにさまざまな適応反応を示す.中でも以下の3つが重要な役割を果たす.すなわち体液量(前負荷)増加,交感神経系やレニン-アンジオテンシン-アルドステロン系活性化による心筋収縮力と血圧の上昇,そして心筋リモデリングである.心室リモデリングとは血行力学的負荷や心筋障害により引き起こされた心臓の構造変化(径,質量,形態)と定義される.その中には運動や妊娠などによる生理的リモデリングも含まれるが,多くの場合は血行動態的異常に伴う病的リモデリングを意味する. 心室リモデリングはおもに,①高血圧や大動脈弁狭窄症などに伴う圧負荷,②弁閉鎖不全症などによる容量負荷,③心筋梗塞,心筋炎などによる心筋障害を原因とする血行動態の変化,により引き起こされることが多い. 圧負荷は求心性リモデリングを引き起こす.このとき,心筋量は増加する場合と増加しない場合があるが,求心性リモデリングは心室壁厚の相対的増加(左室内腔の径に比した壁厚)を特徴とする.求心性肥大に伴い,サルコメアは並列に増加し,個々の心筋細胞は肥大する. 一方,容量負荷や等張性の運動は偏心性の左室肥大を引き起こす.偏心性心肥大は心重量と心臓径の増大を特徴とする.相対的壁厚は変化しないこともあるが偏心性肥大に伴いサルコメアは直列に増加し,個々の心筋細胞径は延長する. 心筋梗塞や心筋炎により心機能の一部が急激に低下した場合,梗塞部のみならず非梗塞部においても求心性および偏心性の混在した肥大が生じることがある.健常部(非梗塞部)だけで心拍出量を維持するために,圧および容量両方の負荷がもたらされるからである.心室リモデリングは心筋梗塞後数時間から発症し,時間とともに進行する.心室リモデリングを発症する症例では,心筋梗塞後4週間をこえて左室の拡大が進行し,徐々に機能不全を呈するようになる.梗塞部は非対称性に菲薄化および拡大を呈する一方,容量負荷による非梗塞部の肥大により心室の形態が高度に変形する(図5-3-3).心筋梗塞後に心室リモデリングを引き起こす要因として,大きな心筋梗塞,Q波心筋梗塞,前壁心筋梗塞,閉塞血管の非開存状態,左室内圧の上昇が考えられている. 当初は代償的な適応機構であったリモデリングがどの段階から予後を悪化させる病的リモデリングに移行するのか,また病態を悪化させる原因がリモデリング自体なのか,あるいはリモデリングを生じさせる誘因なのか,などは今後明らかにすべき問題として残されている.一方,ACE阻害薬やβ遮断薬など心不全患者の予後を改善する薬物治療がリモデリングの進行を遅延させることから,リモデリングの改善は心不全の予後を改善する重要な因子である可能性が高い.現在では不可逆的な病的リモデリングへの進行を予防すべく,心筋梗塞後できるだけ早期にβ遮断薬,アンジオテンシン変換酵素阻害薬の投与が行われている. さまざまな要因により引き起こされる心臓リモデリングであるが,病理学的には共通して心筋壁の拡大,心筋細胞の肥大および間質の線維化が生じている. 心筋細胞の応答は心臓リモデリングにおいて主要な役割を果たす.前負荷が増加,または心筋障害により心筋細胞数が減少した場合,残存した心筋細胞の細胞膜は伸展する.このとき心内圧の増加とあいまって心肥大に関係する遺伝子発現が誘導される.心筋細胞が伸展するに伴い,局所でのアンジオテンシンⅡ,ノルアドレナリン,エンドセリン産生および放出が増加する.これらの液性因子は心肥大の進行および間質の線維化など心筋リモデリングの過程において重要な役割を果たす.アンジオテンシンⅡ,エンドセリンは局所のTNFα,マトリックスメタロプロテイナーゼ(MMP)や酸化ストレスを増加させ,心筋細胞のアポトーシス,ネクローシスを誘導し,心筋細胞の脱落を促す.内腔より受ける圧力の増加はエネルギー代謝バランスの破綻や虚血を引き起こし,それがさらなる心内圧増加を引き起こす悪循環を形成する. 細胞間質,線維芽細胞,コラーゲンなど細胞外マトリックスの役割も重要である.心筋組織はおもにコラーゲンからなる結合組織のネットワークに繋がり支持される心筋細胞からなっている.コラーゲンは間質の線維芽細胞により合成される一方,局所で産生されるMMPなどの酵素により分解されている.コラーゲン合成およびその分解は心臓リモデリングにおいて重要な役割を果たしている.心筋細胞の脱落に対して線維芽細胞の増殖,コラーゲン合成による線維化が促進する一方で,アンジオテンシンⅡ,アルドステロン,炎症性サイトカインはMMP分泌を増加させ,細胞外マトリックスにおいてもリモデリングを引き起こす(Opie 2004).
(2)血管リモデリング
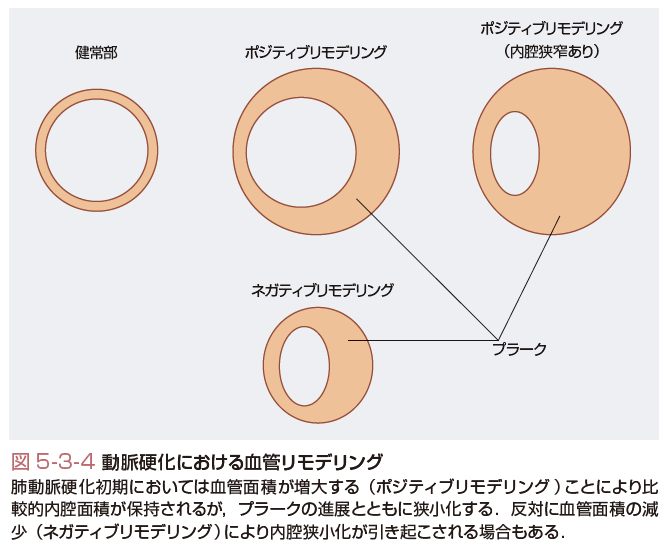
動脈硬化性プラークや,血流のずり応力,血圧の変動,およびカテーテルインターベンションによる血管組織の損傷などが誘因となり,血管径の持続的な拡大または縮小が生じることがあり,血管リモデリングとよばれる.血管リモデリングの存在は剖検における検討から指摘されるようになったが,近年の血管内超音波(intravascular ultrasound:IVUS)検査による同一病変の継時的モニタリングにより直接的に明らかにされた.具体的には,総血管面積を外弾性板より内側の断面積として測定し,健常部の対照血管と比較して増加した場合をポジティブリモデリング,減少した場合をネガティブリモデリングとよぶ.その発症メカニズムとして,血行力学的変化による血管作動物質,増殖因子などによる内皮細胞障害,平滑筋細胞の形質転換に加え,炎症細胞浸潤やMMPによる細胞外基質の再構成を介して血管壁構造の再構築が重要な役割を果たしている. 血管リモデリングは心血管疾患の病態と深く関連している.動脈硬化の初期段階(狭窄度40%以下)においては,動脈硬化プラーク量の増大に伴ってポジティブリモデリングが生じる結果,血管内腔面積(lumen area)の変化はごく軽度にとどまることが多い(Glagov現象,図5-3-4,Glagovら,1987).この段階ではプラーク病変を冠動脈造影で観察しても正常または軽度の血管壁不整を認めるのみである.プラーク増加に伴うポジティブリモデリングは血管内腔面積を維持する代償性作用をもたらしており,その点においては結果として合目的でもある.この考えに基づき,適応反応としてのポジティブリモデリングが起こらない病態を血管機能不全としてとらえる考えも提唱されている. 反対に経カテーテル治療後などに生じるネガティブリモデリングは新生内膜の増大とあいまって,内腔面積の狭小化に寄与する.興味深いことに,スタチン治療などによりプラーク量が減少した場合には,同時にネガティブリモデリングにより総血管面積が減少するため,血管内腔面積の増加は比較的軽度にとどまることが多い(Schbenhagenら,2006). ポジティブリモデリングは血管内腔面積の維持という点では有用である一方で,プラークの不安定化,脆弱性にも関係する. ポジティブリモデリングを伴うプラークは,血管におけるマクロファージ浸潤の増加および炎症マーカー発現の亢進を伴い,また大量のlipid coreを内包しプラーク内血栓を合併することが多い.その一方で平滑筋細胞数が減少し,中膜およびfibrous capが菲薄化する.ポジティブリモデリングは安定狭心症の症例よりも急性冠症候群の患者に多く認められ,不安定狭心症患者において独立した心血管イベントの予測因子であることも確認されている. 反対に,ネガティブリモデリングを伴う病変は石灰化および線維化の程度が高い.動脈のリモデリングの有無は不安定プラークを検出する際の重要なマーカーになる可能性がある.[武田憲彦・永井良三]
■文献
Glagov S, Weisenberg E, et al: Compensatory enlargement of human atherosclerotic coronary arteries. New Engl J Med, 316: 1371-1375, 1987.
Opie LH: Heart Physiology. In Cell to Circulation, 4th ed, Lippincott Williams and Wilkins, Philadelphia, 2004.
Schbenhagen P, Tuzcu EM, et al: Determinants of arterial wall remodeling during lipid-lowering therapy: Serial intravascular ultrasound observations from the Reversal of Atherosclerosis with Aggressive Lipid Lowering Therapy (REVERSAL) trial. Circulation, 113: 2826, 2006.
出典 内科学 第10版内科学 第10版について 情報
Sponsored by ![]()