
内科学 第10版 「病態生理と臨床症状」の解説
病態生理と臨床症状(糖尿病)
a.1型糖尿病の病態生理
1型糖尿病では,膵島のβ細胞が液性免疫と細胞性免疫で傷害され,インスリンの絶対的不足に陥り,インスリン依存状態になることが多い.病理学的には膵島にリンパ球が浸潤し,おもにCD8陽性の細胞傷害性T細胞が特異的に膵β細胞を破壊する.膵島細胞自己抗体,リンパ球活性化,膵島炎によるサイトカイン放出などが膵島破壊に関与する.自己免疫の標的となる膵β細胞特異的な蛋白としてインスリン,神経伝達物質GABAを生合成するグルタミン酸デカルボキシラーゼ(glutamic acid decarboxylase:GAD),チロシンホスファターゼと相同性を有するICA-512/IA-2,インスリン分泌顆粒蛋白などがある.これらの自己抗体には病因的意義は少ないと考えられるが,1型糖尿病の診断の臨床マーカーとして,抗インスリン抗体,抗GAD抗体,抗IA-2抗体が用いられる.
1型糖尿病の疾患感受性にはHLA複合体の遺伝子多型が関与し,MHCクラスⅡ分子やインスリン遺伝子のプロモーター領域など複数の遺伝子が関与する.きわめて短期間にケトアシドーシスに陥る劇症1型糖尿病でもHLA遺伝子の関与が報告されている.環境要因として,コクサッキーウイルスや風疹ウイルスなどのウイルス感染,牛乳蛋白への早期暴露などの関与が想定されている.
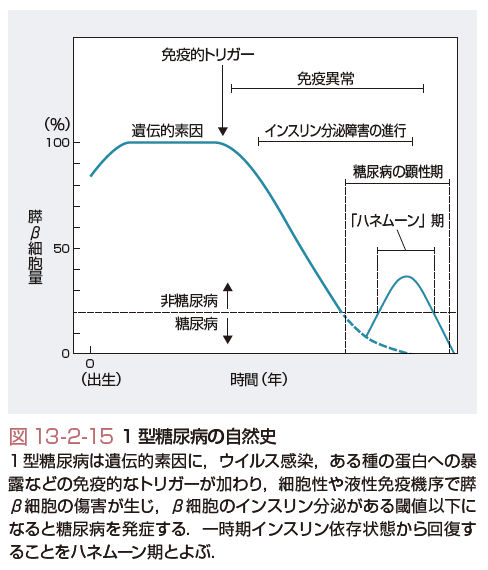
1型糖尿病の自然史を図13-2-15に示す.遺伝的素因を有する個人が,ウイルス感染,ある種の蛋白への暴露などの免疫的トリガーにて,細胞性および液性免疫機序で膵β細胞傷害が生じ,インスリン分泌能が低下する.β細胞のインスリン分泌がある閾値以下になるとインスリン依存状態となり1型糖尿病を発症する.その低下速度が急激な場合は,劇症1型糖尿病,緩徐な場合は緩徐進行性1型糖尿病となる.一時期インスリン依存状態から回復する期間のことをハネムーン期とよぶが,病勢の進行によって再びインスリン依存状態に陥ることが多い.
b.2型糖尿病の病態生理
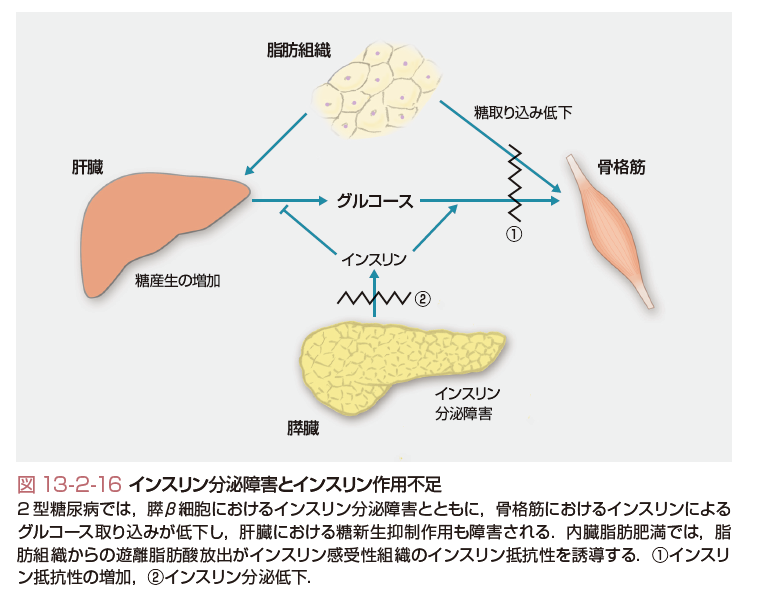
2型糖尿病では,膵β細胞におけるインスリン分泌障害とともに,インスリンによる骨格筋におけるグルコース取り込みが低下し,また,肝臓におけるグルコース新生抑制作用も障害されることから,肝臓からのグルコース放出が増加し血糖値が上昇する.内臓脂肪肥満では,脂肪組織からの遊離脂肪酸放出の増加がインスリン感受性組織のインスリン抵抗性を誘導すると考えられている(図13-2-16).
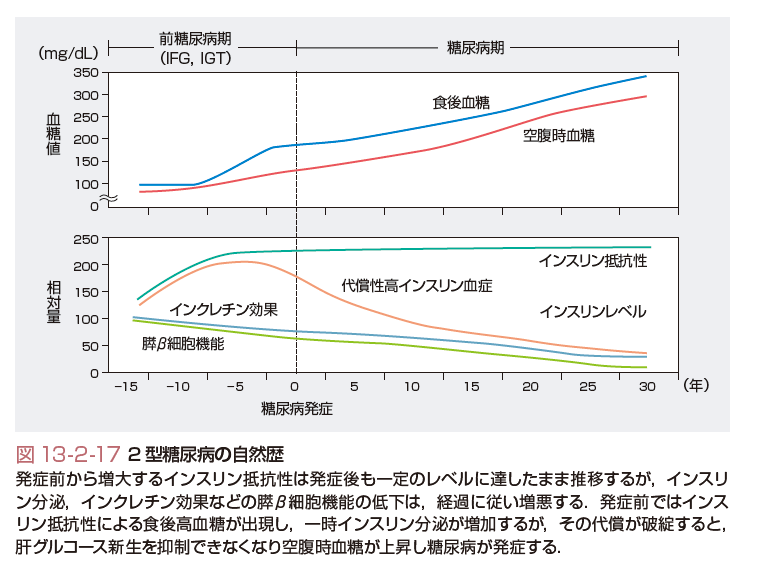
2型糖尿病の自然経過では,発症前から増大するインスリン抵抗性は発症後も一定のレベルに達したまま推移する.一方,インスリン分泌能,インクレチン効果などの膵β細胞機能は疾患の経過に従い低下する.糖尿病発症前には,インスリン抵抗性による食後の高血糖が出現し,一時インスリン分泌が増加するが,代償が破綻するとインスリン分泌が低下し,肝臓のグルコース新生を抑制できなくなり,その結果,空腹時血糖が上昇し,糖尿病を発症する(図13-2-17).
ⅰ)インスリン分泌不全
2型糖尿病ではグルコース負荷によるインスリン分泌が低下し,特にグルコース負荷後の初期分泌反応が障害される.糖尿病ではグルコースによる初期インスリン分泌反応障害が著明であるが,グルカゴンやアルギニンなどに対する反応は比較的保たれていることが特徴である.膵β細胞特異的インスリン受容体欠損マウスやインスリン受容体基質1(IRS-1)欠損マウスが同様にグルコースによるインスリン分泌反応障害を示すことから,膵β細胞におけるインスリン作用不足がグルコース負荷後の初期分泌反応障害に関与することが想定される.また,日本人は遺伝的にインスリン分泌能が欧米人の50~75%程度であるため,肥満などによる軽度のインスリン抵抗性の増大により糖尿病を発症しやすい体質であると推定される.遺伝的な要因として,カリウムチャネル(KCNQ2,KCNJ11,KCNJ15)やインクレチン分泌にかかわる転写因子TCF2L7の遺伝子上の配列の違い(遺伝子多型)が報告されている.最近,膵β細胞量に関して,非糖尿病状態の肥満では,β細胞量や細胞数は増加するが,2型糖尿病ではアポトーシスの増加によりβ細胞量が徐々に減少し,インスリン分泌低下の一因となっている.
ⅱ)インスリン抵抗性
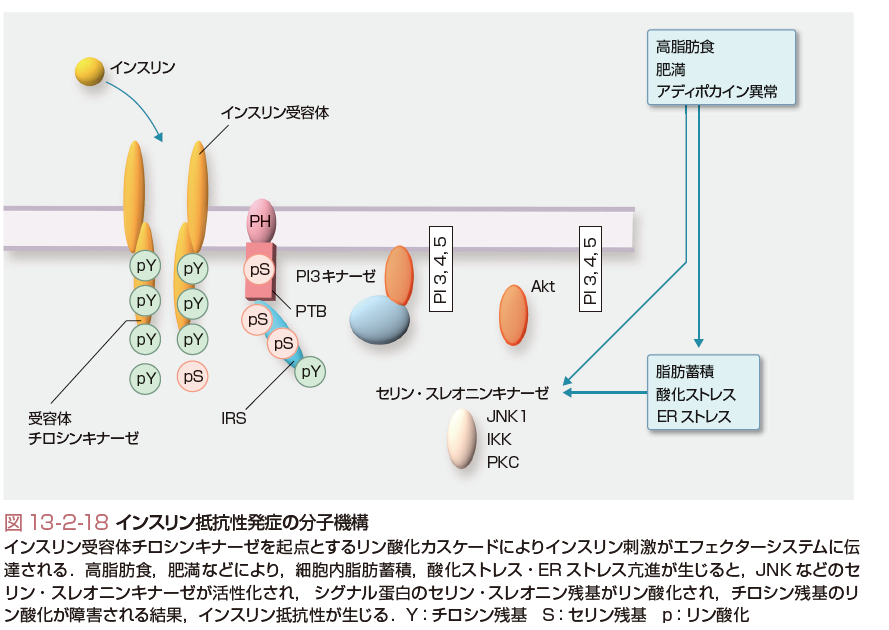
骨格筋,肝臓,脂肪組織などインスリン感受性組織では,インスリンは,細胞膜に存在するインスリン受容体に結合し,受容体チロシンキナーゼの活性化を介してIRS蛋白のチロシン残基をリン酸化する.チロシンリン酸化されたIRS蛋白を介してPI3キナーゼやAktへとリン酸化カスケードが活性化し,糖輸送担体などのエフェクターシステムにインスリン刺激が伝達される.また,インスリンは転写因子Foxoのリン酸化を介して,種々の酵素の遺伝子発現を制御している.高脂肪食,肥満,アディポカイン異常などにより,細胞内脂肪蓄積,酸化ストレスや小胞体(ER)ストレス亢進が生じると,JNK(c-jun N-terminal kinase)などのセリン・スレオニンキナーゼが活性化され,IRS(insulin receptor substrate)蛋白のセリン・スレオニン残基がリン酸化されると,チロシンリン酸化が障害されインスリン抵抗性が生じる(図13-2-18).また,IRS-2蛋白の発現量の減少もインスリン抵抗性にかかわる.
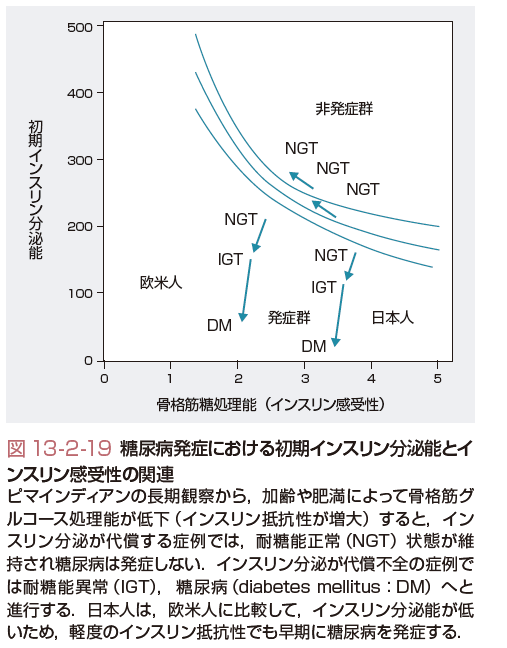
ⅲ)糖尿病発症における初期インスリン分泌能とインスリン感受性の関連(図13-2-19)
ピマインディアンの長期観察から,糖尿病発症における初期インスリン分泌能とインスリン感受性の関連が報告されている.加齢や肥満によって,インスリン感受性が低下し,インスリン抵抗性の増大が生じるが,インスリン分泌の代償性増加が起こる症例では糖尿病は発症しない.しかし,インスリン分泌障害が生じた症例は糖尿病へと進行する.このように,最終的に糖尿病発症はインスリン分泌低下が規定すると考えられている.わが国における糖尿病の急速な増加の背景に,インスリン分泌能がそれほど高くない日本人において,食事の欧米化(肉食中心・高脂肪・高カロリー)や運動不足などによる内臓脂肪蓄積によりもたらされたインスリン抵抗性の増大が大きく関与すると考えられる.
ⅳ)糖毒性と脂肪毒性
インスリン分泌障害およびインスリン抵抗性によって,高血糖状態が持続すると,糖毒性の機構により,さらに,インスリン分泌およびインスリン抵抗性が増大することが知られている.高血糖そのものが,PDX-1などの転写因子を介してインスリン遺伝子発現の障害をきたすことやβ細胞のアポトーシスを促進することが報告されている.
経静脈的グルコース負荷に比し経口グルコース負荷では,同等な血糖上昇にもかかわらず,よりインスリン分泌が刺激されることが「インクレチン効果」として知られている.糖尿病状態では,このインクレチン効果が低下する.その本態は,GIP(glucose-dependent inslinotropic peptide)とGLP(glucagon-like peptide)-1という消化管ホルモンであることが判明し,高血糖状態では,それらの受容体シグナルが減弱していることが示されている.また,高血糖状態は,ヘキソサミン経路の活性化を介して,インスリン抵抗性の増大をきたす.遊離脂肪酸の増加も脂肪毒性を介して,インスリン分泌障害・インスリン抵抗性に関与することが報告されている.
ⅴ)糖代謝異常
【⇨13-2-2)】
2型糖尿病では膵臓からの基礎インスリン分泌が低下している.インスリンはグリコーゲン分解やグルコース新生を抑制しているため,インスリンが欠乏すると肝臓でのグリコーゲン分解やグルコース新生が亢進し,肝グルコース放出が増加し空腹時に著明な高血糖をきたす.インスリン欠乏状態では,食後でも肝臓からのグルコース放出を抑制できず,肝臓,骨格筋および脂肪組織における糖取り込みも低下するため高血糖を生じる.
肝臓では,ピルビン酸脱水素酵素はインスリンにより活性化されるが,インスリン欠乏状態では,この酵素の活性化が抑制され,ピルビン酸からアセチルCoAへの変換が障害され,糖新生の原料となるピルビン酸が増加する.また骨格筋からのアミノ酸(おもにアラニン)の流入も増加するため,インスリンが欠乏した糖尿病では血糖値が高いにもかかわらず,グルコース新生が亢進する.
細胞内に取り込まれたグルコースは通常,グルコース-6-リン酸(G6P),フルクトース-6-リン酸(F6P)となり,解糖系やグリコーゲン合成経路へと代謝される.ヘキソサミン経路は,このF6Pにグルタミンが付加され,グルコサミン-6-リン酸(GlcN6P)となる以降の代謝経路のことを指す.ヘキソサミン経路への糖の流れは全体の2~3%とわずかであるが,高血糖状態では,この経路の亢進が推測される.ヘキソサミン経路の最終産物であるUDP-N-アセチルグルコサミン(UDP-GlcNAc)は,O-GlcNAc transferase(OGT)という酵素の働きで,核や細胞質蛋白のセリン/スレオニン残基の水酸基に結合し,グリコシル化やO-グリケーション(O-GlcNAc)を起こす.最近,インスリンシグナル蛋白のO-グリケーションがシグナル伝達を抑制し,抵抗性に関与することが明らかとなってきた.
ⅵ)脂質代謝異常
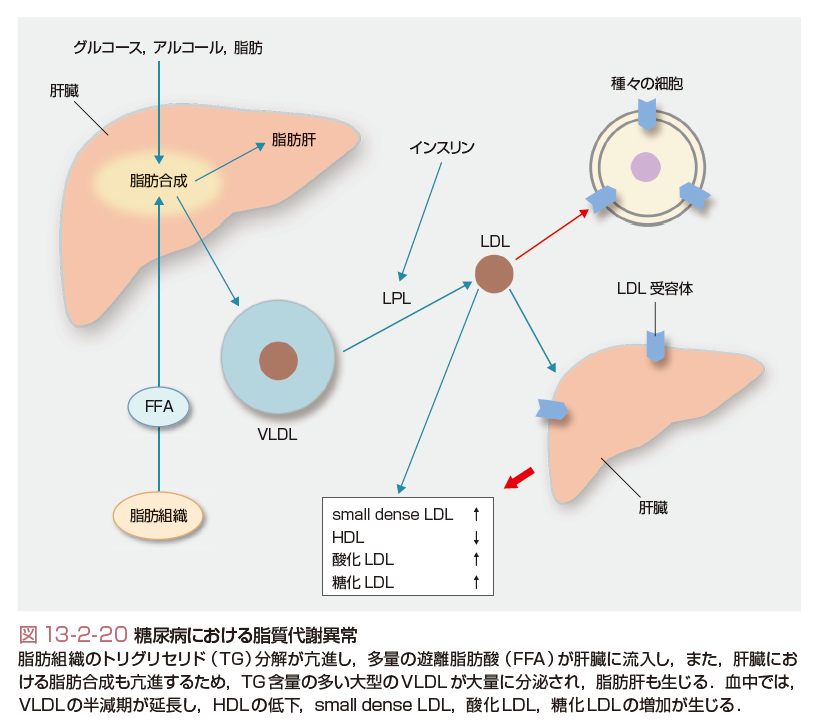
糖尿病状態では,図13-2-20に示すようにインスリン作用の欠乏によりホルモン感受性リパーゼの阻害ができず,脂肪滴内のトリグリセリド(triglyceride:TG)分解が亢進し,遊離脂肪酸(free fatty acid:FFA)が大量に肝臓に流入する.一方,グルコースなどに由来するアセチルCoAから脂肪合成が亢進する.さらに,VLDL(very low density lipoprotein,超低比重リポ蛋白)合成を抑制するインスリン作用も不足するため,TG含量の多い大型のVLDLが大量に分泌される.血中に分泌されたVLDLはリポ蛋白リパーゼ(lipoprotein lipase:LPL)で分解されIDL(intermediate density lipoprotein,中間比重リポ蛋白)になるが,糖尿病ではLPL活性が低下するため,VLDLの半減期が延長する.高インスリン状態では,TG合成は促進するが,VLDL合成は抑制されるため,TGが肝細胞に蓄積し脂肪肝を生じる.血中ではVLDLに多量に含まれるTGが,コレステロールエステル転送蛋白(CETP)によりLDL(low density lipoprotein,低比重リポ蛋白)に転送される.TG含量の多いLDLは,肝トリグリセリドリパーゼにより分解され,small dense LDLが生じる.一方,HDL(high density lipoprotein,高比重リポ蛋白)も大型VLDLからのTG転送により分解が促進され減少する.さらに,高血糖状態では,酸化LDLや糖化LDLが産生され,このような脂質異常の面からも動脈硬化症が進展する.
脂肪肝状態では,グリコーゲン貯蔵や糖新生が阻害され,血中へのグルコース供給能が低下する.また,食後の肝臓での糖取り込み率が低下し食後高血糖を生じる.さらに,夜間の肝臓での糖放出率も増加する.したがって,脂肪肝では,過剰なグルコース流入→インスリン分泌増加→肝臓でのTG合成促進→脂肪肝→食後高血糖や夜間高血糖→インスリン分泌増加という悪循環が形成される.また,血中遊離脂肪酸濃度が上昇すると,プロテインキナーゼC(PKC)活性化,ヘキソサミン代謝経路の活性化,セラミド合成亢進などを介して,インスリン抵抗性を生じると考えられている.
c.糖尿病の臨床症状
ⅰ)高血糖に伴う症状
糖尿病の症状は,高血糖に基づくものと合併症による症状がある.無症状のものから1型糖尿病のように,急速な発症による著明な高血糖やケトアシドーシスのような重篤な症状を示す場合もある.一般的な糖尿病の症状は,高血糖により血清浸透圧が上昇し,圧受容体を介して口渇が生じる.また,尿糖のため多尿となり脱水を生じる.口渇をしのぐため,2~3 Lの水分をとることが多い.多飲,多尿は夜間尿をきたし,特に,高齢者では尿の濃縮力が障害され,夜間の排尿回数が増加する.体重減少や全身倦怠感など非特異的な症状で受診することも多い.体重減少は高血糖による脱水やおもに異化亢進によるものである.また,糖代謝・脂質代謝・蛋白代謝の障害,電解質異常や酸塩基平衡異常により易疲労感などが生じる.
ⅱ)急性合併症に伴う症状
糖尿病性ケトアシドーシス,非ケトン性高浸透圧昏睡など特有の症状がある.【⇨13-2-3)】
ⅲ)慢性合併症に伴う症状
糖尿病性末梢神経障害の症状である足のしびれ,感覚異常を主訴に受診することも多い.神経障害は末梢神経障害と自律神経障害に大別される.末梢神経障害は末梢神経組織内の代謝異常,細小血管病変などを原因とする多発神経障害を主体とするが,神経栄養血管の閉塞性病変に基づく単神経障害や糖尿病性筋萎縮症も認められる.多発神経障害の主症状は両側性の足趾先および足底のしびれ感,知覚低下,異常知覚である.自律神経障害では起立性低血圧,排尿障害,消化管運動障害,勃起障害,無自覚性低血糖など多様な症状が出現する.また,糖尿病網膜症による眼底出血による視力障害や糖尿病腎症によるネフローゼ症候群による浮腫を主訴として来院する症例もある.[前川 聡]
■文献
Bender DA: Overview of metabolism & the provision of metabolic fuels. In: Harper’s Illustrated Biochemistry, 28th ed, pp131-142, McGrawHill Medical, 2009.
Kahn CR, Weir GC, et al eds: Joslin’s Diabetes Mellitus, 14th ed, Lippincott Williams & Wilkins, 2005.
出典 内科学 第10版内科学 第10版について 情報
Sponsored by ![]()