
内科学 第10版 「赤血球膜異常症」の解説
赤血球膜異常症(先天性溶血性貧血)
(1)赤血球膜異常症
a.遺伝性球状赤血球症(hereditary spherocytosis:HS)
概念
先天性溶血性貧血のなかで最も頻度が高く(約70%を占める),約2/3の症例は常染色体優性遺伝,約1/3は常染色体劣性遺伝ないし孤発例である.末梢血中に球状の赤血球(spherocyte)が出現することにより特徴づけられる.
病因
遺伝性球状赤血球症の病因は細胞骨格を構成するアンキリン,バンド3,4.2蛋白やβスペクトリンなどの種々の分子異常による.これらの分子異常によりNa+イオンの透過性が異常に亢進し,Na+の汲み出しのために解糖能が高まり,これから得られるATPを用いたNa+,K+-ATPaseが働いて能動輸送を高め,入り込んでくるNa+を汲み出している.Na+の能動輸送の亢進に伴って膜のリン脂質の喪失がみられ,本症赤血球は小球化する.小球化した赤血球は変形能を失い,脾内を通過できなくなり停滞し,脾の髄索のマクロファージに貪食される.
臨床症状
主症状は貧血・黄疸・脾腫からなる.貧血の程度は症例ごとに異なり,成人になってはじめて発見される例もある.代償性の造血亢進により骨変化がみられることもある.脾腫はほとんどの例で存在し,胆石もしばしば認められる.感染を契機として低形成発作(aplastic crisis)や溶血発作(hemolytic crisis)を起こすと貧血は増悪する.まれに慢性の下腿潰瘍を認める.
診断
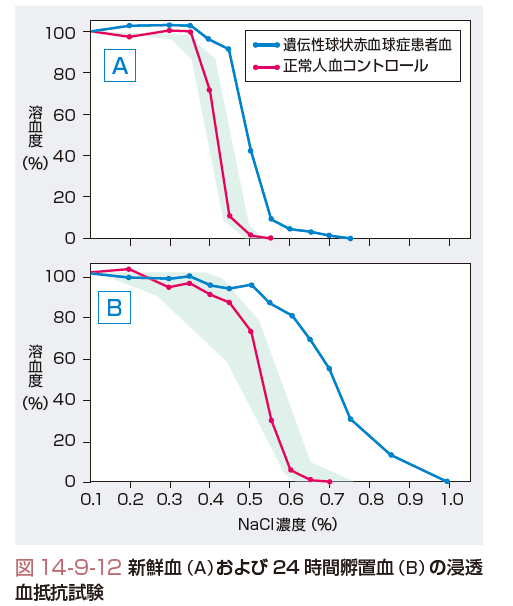
種々の程度の貧血,網赤血球増加,間接ビリルビンの増加,糞尿中ウロビリノーゲンの増加という慢性溶血の一般所見を呈する.溶血の主たる場は血管外であるので,ヘモグロビン尿症は通常みられないが,血清ハプトグロビンは低下する.末梢血塗抹標本では特徴的な小型球状赤血球(microspherocyte;図14-9-11)が認められる.骨髄は赤芽球系過形成像を呈す.溶血性貧血で球状赤血球が存在し,直接抗グロブリン試験陰性で,かつ家族内発生(両親いずれか一方など)がみられればまず遺伝性球状赤血球症を疑う.本症では赤血球浸透圧抵抗は減弱するが,新鮮血で抵抗減弱が証明しにくい場合は,無菌的に37℃,24時間孵置した赤血球を用いた検査(incuvated fragility)では明確になる(図14-9-12).最近は赤血球の形態変化に伴いeosin 5′-maleimide(EMA)結合能が低下する原理を用いて赤血球結合EMAの蛍光強度の低下をフローサイトメトリーで検出する方法も行われている.ほかの溶血性貧血と異なり,本症では平均赤血球ヘモグロビン濃度(mean corpuscular hemoglobin concentration:MCHC)がやや高めの値(35~37%)を呈するので,この値も診断上役立つ.
治療
摘脾により貧血,網赤血球増加や血清ビリルビン値は正常化し,赤血球寿命もほぼ正常に復し,確実に臨床的治癒が得られる.遺伝性球状赤血球症の診断が確実であれば積極的に摘脾を行うべきである.しかし,乳幼児期は摘脾後重症感染症に罹患しやすいので,重症でなければ3歳までは待った方がよい.乳幼児期に摘脾を必要とする場合は,摘脾前に肺炎球菌のワクチンの接種や,摘脾後2年間は抗生物質の経口投与を行う.
b.遺伝性楕円赤血球症(hereditary elliptocytosis:HE)
本症も基本的には常染色体優性遺伝により,病因は遺伝性球状赤血球症と同様にαスペクトリン,βスペクトリン,4.1蛋白,グリコフォリンCやバンド3などの種々な分子異常による.
正常でも楕円赤血球は少数(~数%)は認められるが,遺伝性楕円赤血球症では赤血球の半数以上が楕円形を呈する.遺伝性楕円赤血球症の75%以上の例は無症状であるが,10~15%の例で溶血性貧血を伴う.診断は特徴的な赤血球形態によりなされる.治療としては溶血の著しい症例に対して,遺伝性球状赤血球症と同様に摘脾を行う.
c.その他の赤血球膜異常による遺伝性溶血性貧血
遺伝性口唇状(有口)赤血球症(hereditary stomatocytosis),遺伝性熱変形赤血球症(hereditary pyropoikilocytosis:HPP),遺伝性高赤血球膜ホスファチジルコリン溶血性貧血(hereditary high red cell membrane phosphatidyl choline hemolytic anemia),遺伝性レシチン:コレステロールアシルトランスフェラーゼ欠損症(hereditary lecithin:cholesterol acyltransferase deficiency:LCAT deficiency),β-リポ蛋白欠損症(有棘赤血球症)(abetalipoproteinemia, acanthocytosis),家族性α-リポ蛋白欠損症(familial α-lipoprotein deficiency:Tangier病)などがある.[藤井寿一]
出典 内科学 第10版内科学 第10版について 情報
Sponsored by ![]()