
内科学 第10版 「先天性ムコ多糖症」の解説
先天性ムコ多糖症(その他の代謝異常)
体の構成成分であるムコ多糖の分解経路の1つが障害されるため,各組織の細胞内外に分解されないムコ多糖が経時的に蓄積することによって生じる遺伝性疾患である.
分類
疾患群としては,ライソゾーム病に属する.
原因・病因
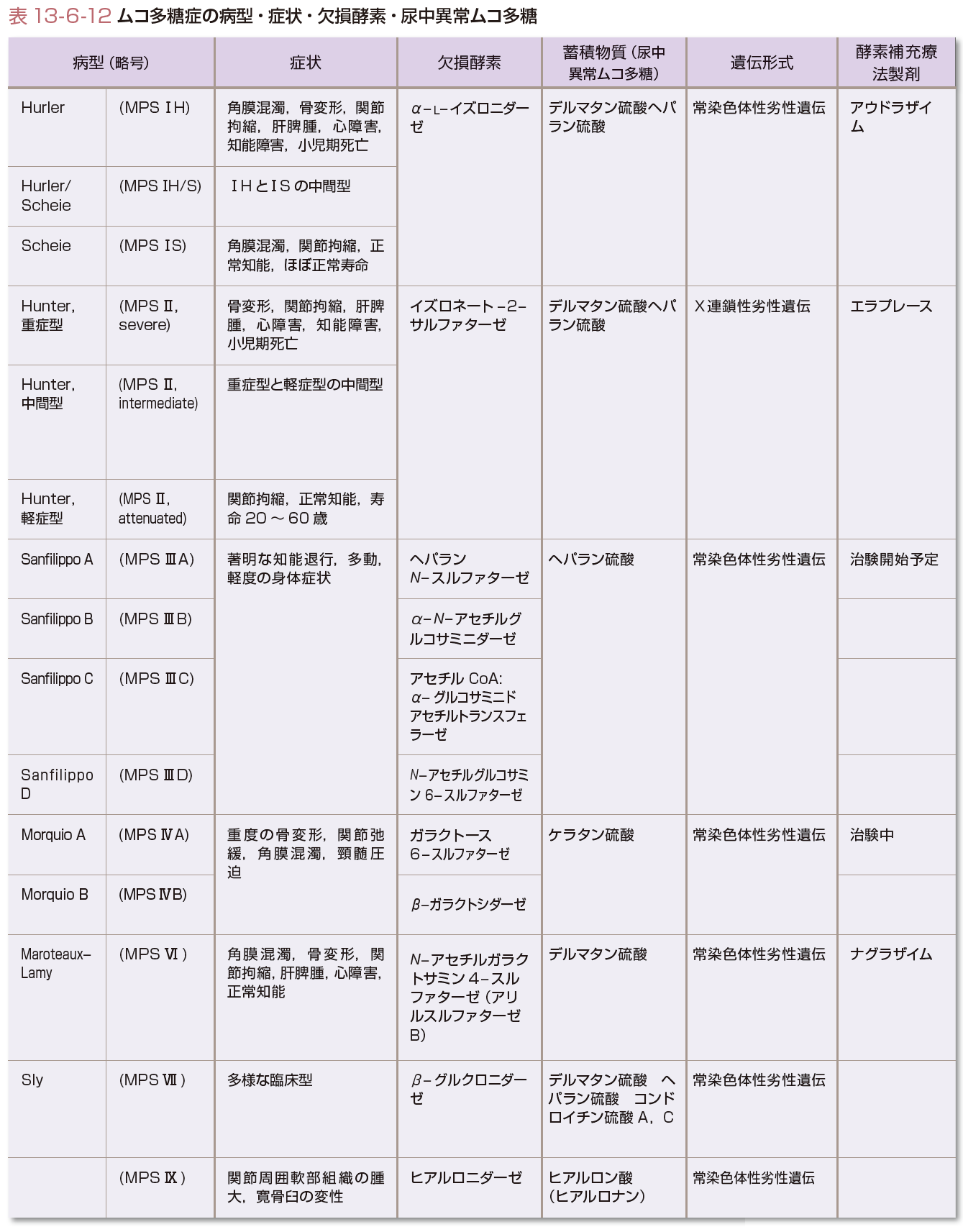
ムコ多糖を分解する酵素の遺伝子異常により表13-6-12に示す酵素活性が低下することにより起こる.
疫学
ムコ多糖症Ⅱ型だけがX-連鎖性遺伝であり,ほかはすべて常染色体性劣性遺伝の疾患である.患者の頻度は,日本を含め東アジア地域ではムコ多糖症Ⅱ型が最も多く,ムコ多糖症全体の半数以上を占める.ムコ多糖症全体の発症率は,およそ10万人に1人である.
病理
光学顕微鏡では,ライソゾーム内に非分解産物が蓄積することや非代謝産物をマクロファージが貪食することによる特徴的な所見が認められる.また,間質マトリックスはムコ多糖が多量に蓄積し,トルイジンブルーやアルシアンブルーに濃染する.
病態生理
ムコ多糖は,結合組織の構築成分として常に新生と分解を繰り返している.分解は,細胞のライソゾームにおいて加水分解酵素により行われる.この酵素の1つが遺伝的に欠損しているために,ムコ多糖症(mucopolysaccharidosis)とよばれる一群の疾患が生じる.表13-6-12に,欠損酵素と分解が障害され蓄積するムコ多糖の種類および各病型の臨床症状を示す.分解されないムコ多糖が経時的に蓄積するため,症状は次第に現れて進行する.同じ酵素欠損症であっても,その遺伝子変異の有様によって疾患重症度が異なる.点変異によりできた変異酵素蛋白が比較的酵素活性を残存している場合にはゆっくりと症状が現れて徐々に進行するが,塩基の欠失や挿入などで大きく酵素活性が損なわれる場合には,早期に症状が現れて急激に進行する.
臨床症状
表13-6-12に簡単に示すように,病型により様相が異なる.
1)ムコ多糖症Ⅰ型:
下記に記すように幅広い臨床型を示す.①Hurler病は,ムコ多糖症全体の中でも重症であり,ほとんどは10歳頃までに死亡する.6カ月から2歳頃に,肝脾腫,骨変形,粗な顔貌,関節拘縮,角膜混濁,巨舌などに気づかれる.乳児期より,臍ヘルニア,鼠径ヘルニアが認められることが多い.発達は,2歳から4歳をピークとしてその後,後退する.上気道感染,中耳炎が頻回で,騒音性の呼吸,慢性の多量の鼻汁が認められる.骨X線では,頭蓋骨の肥厚,椎骨前縁の形成不全,骨盤骨および大腿骨骨頭の形成は不良で,外反股を示す.肋骨は椎骨側が細く胸骨側は幅広くなりオール状の変形を示す.②Hurler/Scheie病は,Hurler病とScheie病の中間の臨床表現型を示すものを指す.知能障害はないか,ごくわずかである.症状の発現は,3歳から6歳の間に気づかれ進行する.10歳代半ばまでに,角膜混濁,関節拘縮,聴力障害,心臓弁の障害のため,生活に支障が起こる.③Scheie病は,5歳以降に,関節拘縮,大動脈弁狭窄・逆流,角膜混濁,肝脾腫などが起こってくる.10歳から20歳の間に診断されることが多い.粗な顔貌,鷲手,凹足,外反股,手根管症候群などを認める.知能は正常である.角膜混濁以外に緑内障,網膜変性も起こり,深刻な視力障害をきたす.閉塞性呼吸障害による睡眠時無呼吸をみる.大動脈弁,僧帽弁にムコ多糖が蓄積するため狭窄・逆流が起こる.壮年期にまで達することができる.
2)ムコ多糖症Ⅱ型(Hunter病)
重症型,軽症型に分けられる.中間型も存在する.ムコ多糖症I型と類似の症状を呈するが,角膜混濁はみられない.遺伝形式は,ほかのムコ多糖症が常染色体性劣性遺伝形式であるのと異なり,X連鎖性劣性遺伝形式である.①ムコ多糖症Ⅱ型重症型は,2〜4歳の頃に,粗な顔貌,骨変形,関節拘縮,肝脾腫,知能障害が始まり進行する.Hurler病と似ているが,進行はややゆるやかで角膜混濁がない.臍ヘルニア,鼠径ヘルニアも多くみられる.幼児期の体格はむしろ大きいが,5~6歳以降に発育が停止する.10〜15歳で死亡する.おもな死因は,閉塞性の呼吸障害や心臓弁の障害,心筋肥大,肺高血圧,冠動脈の狭小化による心不全である.②ムコ多糖症Ⅱ型軽症型は,5〜7歳の頃に,骨変形,関節拘縮に気づかれる.重症型と同様の症状が出現し進行するが緩徐であり,知能障害はない.聴力障害,手根管症候群,関節拘縮のため,生活に支障が起こる.寿命は20歳代~50歳代である.
3)ムコ多糖症Ⅲ型(Sanfilippo症候群)
表13-6-111に示すように4つの亜型に分けられる.白人においてはⅢA型が最も多く,ⅢB型がそれにつぐ.日本人では,ⅢB型の方が多い.ⅢC型,ⅢD型は,どちらもまれである. 臨床症状としては,いずれの亜型においても大変似かよっており,重症の中枢神経変性症状が特徴的で,身体症状は軽度である. 2〜6歳頃に症状が発現する.多動,乱暴な行動,発達遅滞,粗い毛,多毛が認められる.中枢神経変性症状が急速に進行し,7~8歳までに言語は消失する.10歳代になると,睡眠障害,肝脾腫,痙攣発作がみられ,周囲とのコンタクトも消失する.Sanfilippo症候群は,ムコ多糖症に特徴的な粗な顔貌や関節・骨の変形は軽度である.10歳代で寝たきりとなり,多くは20歳代頃に呼吸器感染症などで死亡する.
4)ムコ多糖症Ⅳ型(Morquio病)
ⅣA型とⅣB型がある(表13-6-12).ⅣA型が圧倒的に多い.
臨床的には,骨の変形が特徴的で,ムコ多糖症の中で最も強い骨の変形を示す.知能は障害されない.角膜混濁がある.ⅣB型は,比較的軽症とされている.
幼児期になって,外反股,四肢の変形,亀背,低身長,短頸,短躯,角膜混濁に気づかれる.靱帯が弛緩し,関節可動域は増大するが,大きな関節では,骨の変形の影響で減少する.歯状突起の形成不全と靱帯の弛緩に加えて,頸髄周囲の硬膜が肥厚し,C1,C2レベルで後方より頸髄が圧迫される.同様のことは,IH型,Ⅵ型,Ⅶ型にもみられるが,Ⅳ型が最も重度である.程度が進むと上下肢の麻痺や呼吸障害をきたす.
5)ムコ多糖症Ⅵ型(Maroteaux-Lamy病):
Hurler病に似た身体所見を示すが,知能は障害されない.重症型,軽症型がある.
重症型の身体所見はHurler病とよく似ている.骨の変形や関節拘縮が,すでに1歳頃より認められる.臍ヘルニア,鼠径ヘルニアも多くみられる.角膜混濁,肝脾腫,皮膚の硬化,鷲手,手根幹症候群,腰椎前弯,大動脈弁・僧帽弁の肥厚がみられ,10~20歳代に心不全により死亡する.骨X線もHurler病とよく似る.
6)ムコ多糖症Ⅶ型(Sly病)
最重症型の新生児型では,胎児水腫として認められ,胎児期または乳児期早期に死亡する.重症型は,Hurler病に似ており,3歳頃までに種々の症状が現れる.軽症型は,4歳以降に症状が現れ,骨の変形がおもな症状で,知能障害も角膜混濁も認められない.病型や,Hunter病の中間型のような病型が存在する.日本人の報告としては,後者が多い.末梢血顆粒球中に異常な封入体がみられることが特徴であるが全例ではない.
7)ムコ多糖症Ⅸ型:
症状は非常に軽微である.関節周囲の軟部組織腫瘤,軽度の低身長,寛骨臼の不整を認めるが神経症状,臓器症状はない.
検査成績
1)一般血液尿検査:
特異的異常値を示す疾患は,ほとんどない.肝腫大によりAST,ALTの軽度上昇,骨症状の進行のためALPの上昇を認める.
2)放射線検査:
a)単純X線:椎体の楔状変形,肋骨のオール状変形など特徴的な骨変形が認められる.診断の手がかりとして最も重要である. b)MRI:血管周囲腔の拡大が認められる.脳梁部におけるものが特徴的である.Ⅰ型,Ⅱ型,Ⅵ型で高頻度に認められるが,Ⅲ型やⅣ型では頻度が低い.Ⅱ型の重症型では,大槽の拡大が高頻度にみられる.また,Ⅰ型,Ⅳ型では上部頸髄の圧迫像がしばしばみられる.
3)生理検査:
a)呼吸機能検査:閉塞性,拘束性の呼吸障害を認める.巨舌やアデノイド,声門狭窄による気道の狭小と,胸郭の動きが悪いことが原因である. b)心臓超音波検査:大動脈弁と僧房弁がおもな病変部位である.初期は弁尖が肥厚して輝度が上昇する.弁の動きは次第にしなやかさを失い棍棒のようにみえる.開放制限と閉鎖不全が同時に起こってくる.大動脈弁においては,弁輪部が拡大して逆流はより顕著になる.心筋においては求心性の肥厚が起こり,駆出率は保たれるものの拡張障害が起こる.逆流による力学的負荷が長期に及ぶことにより心拡大が起こり,心筋の肥厚はむしろ減少して駆出率も低下してくる.心不全となり死因となる.
4)眼科的検査:
Ⅰ型,Ⅳ型,Ⅵ型,Ⅶ型では角膜混濁を合併する.程度は,およそⅠ型,Ⅵ型,Ⅳ型/Ⅶ型の順に重症である.病型を問わず,症例により眼圧の亢進が起こる.長期に持続すると網膜変性をきたす.
5)耳鼻咽喉科的検査:
聴力検査で伝音性と感音性の混合性の難聴を認める.滲出性中耳炎,アデノイドを認める.声帯が肥厚,硬化するため嗄声となる.
診断・鑑別診断
特徴的な骨の変形によりムコ多糖症が疑われる.尿中のムコ多糖分析を行い異常なムコ多糖の排泄を認めたならば,末梢白血球またはリンパ球を用いて酵素活性を測定することにより確定診断を行う(表13-6-12).よく似た骨の変形を呈する疾患に,ムコリピドーシス,GM1-ガングリオシドーシスがある.これらは,尿中に異常なムコ多糖の排泄を認めない.
経過・予後
ほぼすべての疾患は,進行性であり,病状が進行して死に至る.寿命の長さは,病型,重症度により異なり,小児期に死亡するものから50歳以上のものまである.死因としては,Ⅰ型,Ⅱ型の軽症型では心不全が多く,重症型では呼吸器感染症が多い.Ⅲ型では中枢神経の荒廃による呼吸停止,Ⅳ型では頸髄圧迫による呼吸不全・呼吸停止が多い.
治療
1)酵素補充療法:
Ⅰ型,Ⅱ型,Ⅵ型で酵素製剤による酵素補充療法が可能である(表13-6-12).効果は,肝臓,皮膚,毛髪,舌,気道粘膜には良好であるが,脳神経,骨,心臓弁には乏しい.脳神経に対する効果を得るために,髄腔内投与の臨床治験がⅡ型において進められており,Ⅲ型についても計画されている.
2)造血幹細胞移植:
多くの病型で試みられたが,効果の程度,臓器種は酵素補充療法とほぼ等しい.したがって,脳神経病変が主たる症状のⅢ型や骨病変が主たる症状のⅣ型には推奨されないが,Ⅰ型,Ⅱ型,Ⅵ型でよい条件の骨髄ドナーや臍帯血があり移植合併症が少ないことが推定される症例では,早期に行うことが望ましい.
3)遺伝子治療:
Ⅰ型において自己の骨髄細胞に正常遺伝子を導入し,それを患者に戻すex vivoの遺伝子治療の臨床治験が計画されている.ⅢA型では,ベクターに組み込んだ遺伝子を直接髄腔内に入れるin vivoの遺伝子治療の臨床治験がフランスで進行中である.
予防・遺伝カウンセリング
効果的な治療法がないことから,遺伝カウンセリングを行って,出生前診断により罹患児出生を防ぐという選択をされることもある.[田中あけみ]
出典 内科学 第10版内科学 第10版について 情報
Sponserd by ![]()