
内科学 第10版 「先天性ミオパチー」の解説
先天性ミオパチー(筋疾患)
骨格筋は,発生・分化や再生のように正常筋構造を形成する過程と筋細胞壊死などの崩壊とのバランスを保ちながら正常な収縮活動を担っている.先天性ミオパチーは筋の発生・分化や再生の障害が本態であり,先天性筋ジストロフィは筋の崩壊の方に病気の本態がある.先天性ミオパチーは,①特徴的な筋細胞内の構造的変化,②新生児ないしは乳幼児期からの筋力低下での発症,③非進行性(緩徐進行性),④遺伝性,の4つの特徴をもつ病気の総称である.
分類
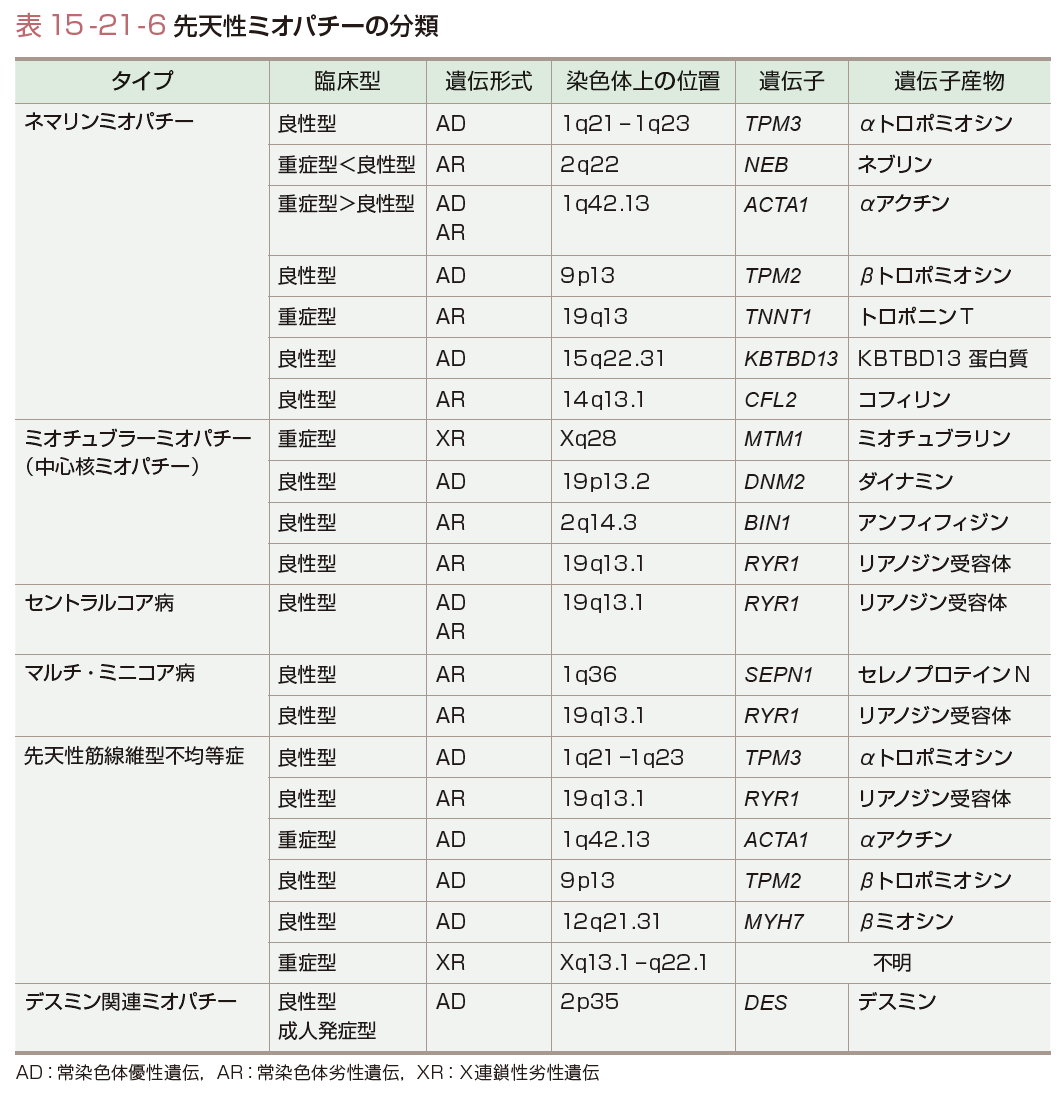
特徴的な筋細胞内の構造異常に基づいて分類されている(表15-21-6).
原因・病因
表15-21-6のように,ネマリンミオパチーでは,筋原線維の収縮蛋白質であるαアクチンや骨格蛋白質であるネブリンの遺伝子変異が同定され,また収縮を調節する蛋白質群(αトロポミオシン,βトロポミオシン,トロポニンTなど)の遺伝子変異も同定されている.また,ミオチュブラーミオパチーでは,X連鎖性の遺伝形式をとる最重症型でMTM1遺伝子の変異が同定されている.遺伝子産物であるミオチュブラリンは,チロシン脱リン酸酵素の1つである.セントラルコア病では,リアノジン受容体遺伝子(RYR1)の変異が報告されている.104個のエクソンからなるこの巨大遺伝子の産物は筋小胞体膜上に位置し,カルシウムチャネルの開閉に関係している.同じ遺伝子の変異は全身麻酔薬投与で起こる悪性高熱と関連する.RYR1遺伝子の変異は,セントラルコア病以外に,ミオチュブラーミオパチー,マルチ・ミニコア病,先天性筋線維型不均等症など複数の表現型をきたすことが判明し,これら構造異常によるミオパチーの病因の共通性が明らかになった.
疫学
筋ジストロフィに比較してまれな病気であり,厚生労働省小児慢性特定疾患治療研究事業での患者登録数は年間20人程度である.
病理
先天性ミオパチーの共通の筋病理所見として,萎縮したタイプ1線維(赤筋)が55%以上の優位に存在し,タイプ2線維(白筋)が欠損もしくは部分欠損し,また未分化な筋線維を認める.この共通所見に加え,存在している特徴的な構造異常をもって診断名となり,共通所見だけの場合は先天性筋線維型不均等症という診断名がつけられる.代表的な病気で認める骨格筋の構造異常を図15-21-12に示す.
病態生理
先天性ミオパチーのそれぞれの病気は,構造異常をもつ筋細胞が直接臨床症状という臓器レベルの病態を反映しているのではない.たとえば,ネマリンミオパチーにおいてはネマリン小体の大きさや出現頻度などと症状との相関は認められない.筋力低下という臨床症状を引き起こしている基本病態は,筋線維数の絶対数,すなわち筋量の減少である.
筋細胞の分化・成熟には,筋の幹細胞自体から発現される筋分化因子以外に,神経性の因子(末梢神経側から与えられる活動電位など)が必要なことが知られている.また,腱の切断,局所的持続的な収縮刺激,脱神経後の神経再支配過程,逆に関節固定や完全な脱神経でも筋細胞内にコアなどの形態変化が認められることがあることから,先天性ミオパチーにおいても神経性因子の関与を考える必要がある.
臨床症状
先天性ミオパチーは,いずれも臨床症候に共通点があり,病歴,遺伝形式,身体的所見だけでは,各病気の鑑別は困難である.臨床経過から重症型,良性型,成人発症型に分けられるが,セントラルコア病では重症型は認められていない.
重症型は,新生児期から呼吸障害,嚥下障害を認め,ほとんどが1歳以下で死亡する.良性型は最も多い型で,乳児期早期からの筋緊張低下と筋力低下があり,定頸,寝返り,処女歩行などの運動発達の遅れで気づかれる.その後,転びやすい,階段昇降が困難である,走るのが遅いなどの症状が続く.体格は細く,顔も細長いことが多い.顔面筋罹患があるため口蓋の発達が不十分で高口蓋となり,表情も乏しい.尖足などの関節拘縮や側弯などの姿勢異常も認める.知的発達は正常であることがほとんどである.
各病気の特異的な症候として,ネマリンミオパチーの良性型では比較的呼吸筋が侵されやすいという特徴がある.セントラルコア病では,顔面筋罹患が少なく高口蓋もないことがある.しかし悪性高熱の合併をみることがあるので,診断がついた後の全身麻酔時には特に注意が必要である.ミオチュブラーミオパチーの良性型では,比較的顔面筋が侵されやすく,上眼瞼下垂や眼球運動障害を認めることがある.また約10%程度に知的発達障害やてんかんを合併することがある.ミオチュブラーミオパチーの重症型は,新生児期から重篤な呼吸障害を伴い致死的である.先天性筋線維型不均等症でも,約30%に中枢神経系の異常を認める.
検査成績
筋ジストロフィと異なり,血清クレアチンキナーゼ,アルドラーゼなどの筋逸脱酵素の値は正常もしくは軽度に上昇するのみである.筋電図は筋原性変化を示す.筋CTは全身の筋量を評価するのに有用である.
診断
確定診断には,筋生検が必要である.
合併症
新生児期から発症する重症型の場合は,自発呼吸が出ずに生直後から人工呼吸管理が必要になる.良性型でも進行すると呼吸筋が障害され,人工呼吸管理が必要となることが多い.
ミオチュブラーミオパチーや先天性筋線維型不均等症などでは,知的発達障害やてんかんが合併することがある.デスミン関連ミオパチーでは,骨格筋以外に心筋も侵される.
経過・予後
筋症状は非進行性であったり,緩徐進行性であったりするが,ときに呼吸障害が急速に進むことがあるので注意を要する.重症型は,中枢神経症状がなければ,呼吸管理とその合併症である呼吸器感染症への対処の善し悪しによってその予後が決まるといってよい.
良性型は,粗大運動発達の遅れが主訴で診断に至ることがふつうであり,その後も発達が遅れ,ついには次第に運動機能が低下してくる.
治療・予防・リハビリテーション
原病の治療として有効なものはなく,合併症の予防やその早期治療が目標となる.患者がもつ運動機能に対応した,適切な補助器具の使用や関節拘縮の防止のためのリハビリテーションが必要になる.[後藤雄一]
■文献
埜中征哉:先天性ミオパチー.臨床のための筋病理 第4版,pp110-131,日本医事新報社,東京,2011.
出典 内科学 第10版内科学 第10版について 情報
Sponsored by ![]()