
内科学 第10版 「Marfan症候群」の解説
Marfan症候群(先天性結合織疾患に伴う心血管病変)
概念
Marfan症候群は1896年にMarfanにより報告された,大動脈,骨格,眼,肺,皮膚,硬膜などの全身の結合組織が脆弱になる,遺伝性疾患である.結合組織が脆弱になることにより,長い四肢,くも状指趾,関節過伸展,長頭型頭蓋などの骨格異常,水晶体亜脱臼,大動脈瘤や大動脈解離,僧帽弁逸脱症,自然気胸などをきたす.
病因
常染色体優性遺伝を呈し,約75%は家族性であり,約25%は突然変異である.病因遺伝子は,fibrillin-1(FBN1)遺伝子が主要なものであるが,トランスフォーミング増殖因子β2型受容体(TGFBR2)遺伝子や,TGFBR1遺伝子が病因の家系も報告されている.
病理
大動脈は,エラスチン線維の断片化と配列の消失,平滑筋細胞の減少,中膜の細胞間へのコラーゲンおよびムコ多糖類の沈着により特徴づけられる.この特徴により囊胞性中膜変性と記載される.弁へのムコ多糖類の沈着が弁膜の肥厚を引き起こす場合もある.
臨床症状
心臓血管系合併症で,最も罹患率と死亡率の高いものは大動脈基部の拡張(annuloaortic ectasia)である.大動脈瘤破裂や大動脈解離により突然死をきたすことがある.大動脈拡張に伴う大動脈弁閉鎖不全により心不全や,大動脈基部の解離による冠動脈入口部閉塞による心筋梗塞を発症する場合もある.大動脈解離では,末梢臓器循環不全を呈することがある.また僧帽弁逸脱や閉鎖不全,肺動脈拡張などがみられる.
骨格系の所見としては,両手を広げた長さ(arm span)/身長比が1.05以上,手指徴候(wrist sign,手首を握ると母指が第5指と重なる)および母指徴候(thumb sign,母指を中に入れて拳をつくると母指が尺側から突き出る),鳩胸,漏斗胸などがみられる.
診断
Ghent診断基準では,心臓血管系の主要基準に大動脈基部の拡張と上行大動脈解離,併発症状に僧帽弁逸脱症, 40 歳未満での肺動脈拡張や僧帽弁輪石灰化,その他の大動脈の拡張もしくは解離があげられている.また2010年に改訂されたGhent診断基準では大動脈基部径の評価,水晶体転位の有無,家族歴の有無,遺伝子診断の結果などが重要視されている.
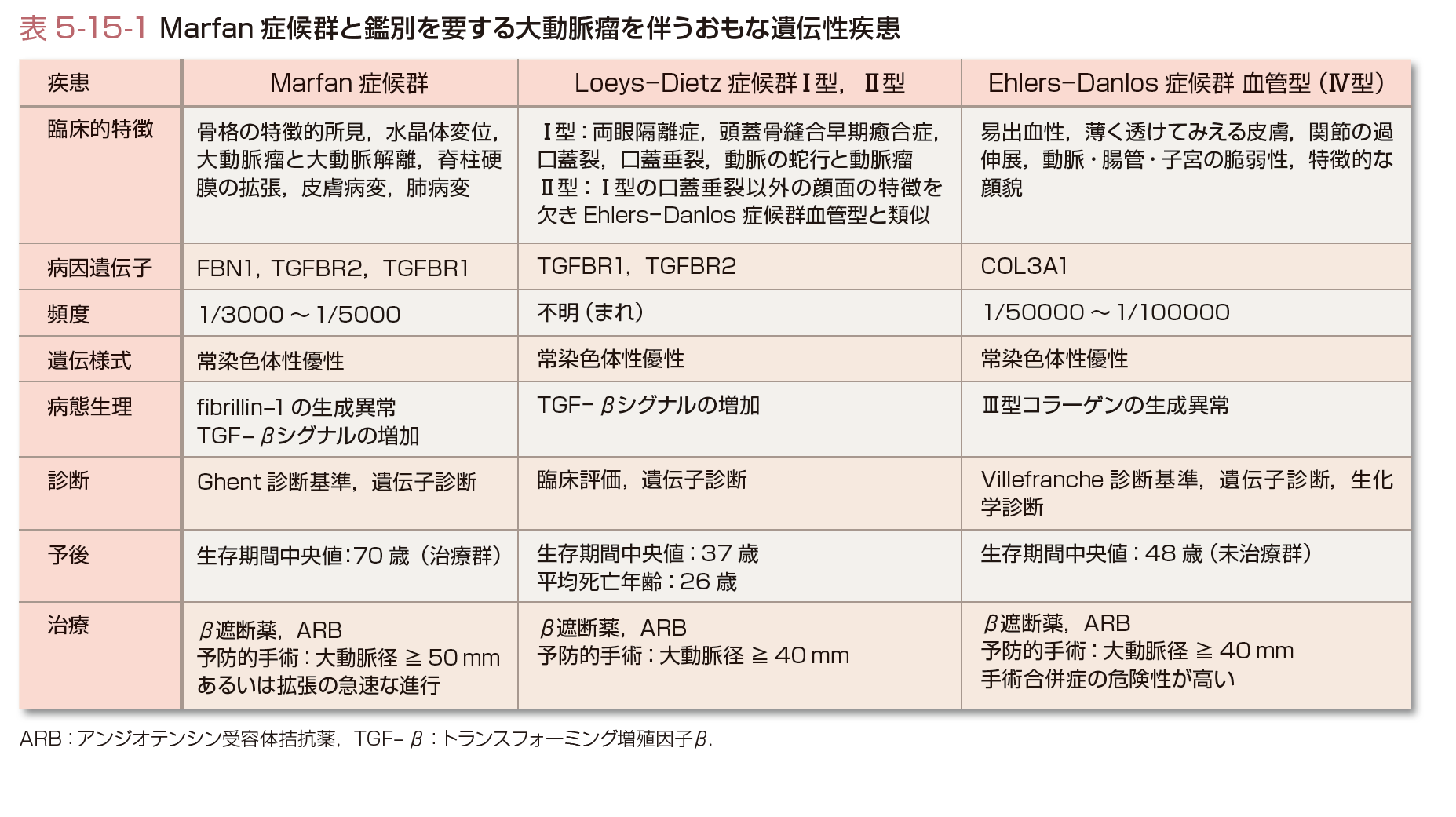
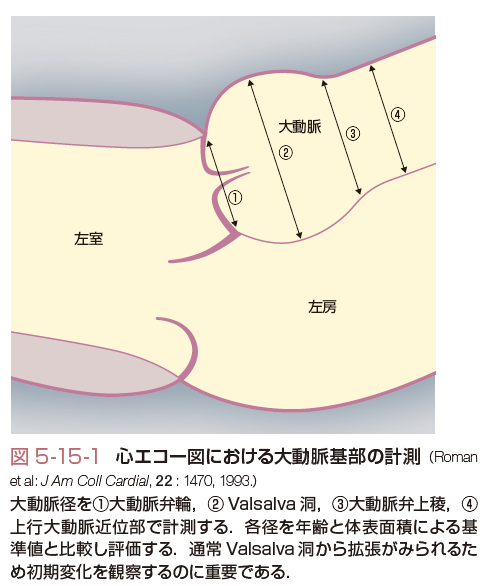
X線,心電図,エコー,CT,MRI,などで大動脈基部の拡大,大動脈解離の有無,大動脈弁,僧帽弁の異常の評価を行う.心エコーによる大動脈基部の評価について示す(図5-15-1).表5-15-1にMarfan症候群と鑑別を要する大動脈瘤を伴うおもな遺伝性疾患の特徴を記載する.
経過・予後
大動脈解離の危険因子として,5 cm以上の大動脈径,Valsalva洞をこえた大動脈の拡張,急激な拡張率(年5% 以上,もしくは成人で年1.5 mm 以上),大動脈解離の家族歴があげられる.したがって年に1回は心エコーを含む評価を行うべきである.妊娠時には,大動脈径が4 cmをこえると大動脈解離の危険性が増す. 医学的介入により,心血管合併症による死亡率は1972年には70%であったのに対し1995年には48%と低下し,死亡平均年齢は1972年は32±16歳に対して1998年には45±17歳と上昇している.
治療
大動脈瘤,解離に対しては,降圧ならびに心拍数減少の目的にて,β遮断薬による薬物療法が行われる.大動脈の拡張がみられる場合には,年齢を問わずに行う.最近のTGF-βの過剰活性化の知見から,TGF-βの機能を抑制するアンジオテンシン受容体拮抗薬の投与が試みられている. 大動脈瘤,大動脈解離に対しては,人工血管置換術を行う.Valsalva洞の大動脈径が5 cmをこえた場合には,予防的な大動脈基部の手術を考慮すべきである.予防的大動脈置換術は大動脈径が4 cm未満の場合により効果的と考えられる.[阿南隆一郎・鄭 忠和]
■文献
Prockop DJ, Bateman JF: Heritable disorders of connective tissue. In: Harrison’s Principles of Internal Medicine, 18th ed (Longo DL, et al ed), pp3204-3214, McGraw-Hill, New York, 2012.
Dean JC: Marfan syndrome: clinical diagnosis and management.Eur J Hum Genet, 15: 724-33, 2007.
Cañadas V, et al: Marfan syndrome. Part 1 and 2: pathophysiology and diagnosis. Nat Rev Cardiol, 7: 256-276, 2010.
Loeys BL, et al: Aneurysm syndromes caused by mutations in the TGF-β receptor. N Engl J Med, 355
: 788-798, 2006.
Marfan症候群(先天性結合組織疾患)
頻度
5000出生に1人の割合で存在するといわれ,地域差や人種差は認められていない.75%の症例はそのどちらかの親がMarfan症候群であるが,25%は遺伝性ではなく突然変異により発症する.常染色体優性遺伝であるが男性の方が重症である.多くは30~40歳前に心血管系の異常で死亡する.
臨床症状
典型的には手足が長く高身長,眼の水晶体の偏位,胸郭の変形,大動脈解離などが有名であるが,これらを含めおもな臨床症状を以下に示す.
大動脈基部の拡張や大動脈弁逆流症が多くの症例で認められ,死因としても重要である.拡張は大動脈基部のみならず胸部・腹部大動脈にも存在することがあり,やがて大動脈解離を引き起こす.若年者(40歳以下)で大動脈解離を発症したものの半数はMarfan症候群であるといわれている.また,僧帽弁逸脱症を認めることもある.
長管骨が長いため高身長であり,手指も長くくも状指(arachnocdactyly)とよばれる.両腕を広げた長さは自らの身長よりも長くなる.また,自らの手首を反対の手で握った場合,反対側の親指と第5指が重なる手首徴候(wrist sign)や親指を中にして握り拳をつくった場合に親指の先が第5指側から出てしまう親指徴候(thumb sign)などを認める.鳩胸あるいは逆の漏斗胸や脊椎の側後弯,偏平足や関節の過伸展も認められる. 眼症状として水晶体偏位(ectopia lentis)を50%以上の症例で認める.水晶体を支持する組織の脆弱性のために上方耳側に偏移することが多い.強度の近視から網膜剥離を生じる場合がある. 腰仙椎部の硬膜拡張が多くの症例で存在し,MRIでの検出感度が高い.
診断
2010年に改訂Ghent分類基準が発表された.これは身体的特徴(骨格系),心血管系,眼症状,家族歴・遺伝子異常を総合して判断するが,家族歴がない場合(de novoの遺伝子変異)や軽症例では鑑別すべき疾患が多くあり,困難である.
治療
疾患の性質上,保存的治療が大きな部分を占める.適度の運動は推奨されるが,過度の身体的負荷を避け,心血管系への負担を軽減しなければならない.超音波検査により大動脈(基部)の拡張の程度をモニターすることにより,必要に応じて行う外科的手術に備える.特に大動脈基部の径が50 mmをこえると外科的治療が推奨される.薬物としては血管系への保護作用がある,アンジオテンシン変換酵素拮抗薬,アンジオテンシン受容体拮抗薬,あるいはカルシウム拮抗薬などが試されている.特にβ遮断薬は禁忌が存在しない限り使用することが推奨されている.[簑田清次]
出典 内科学 第10版内科学 第10版について 情報
Sponsored by ![]()