
内科学 第10版 の解説
心臓の発生と先天性心疾患(先天性心疾患)
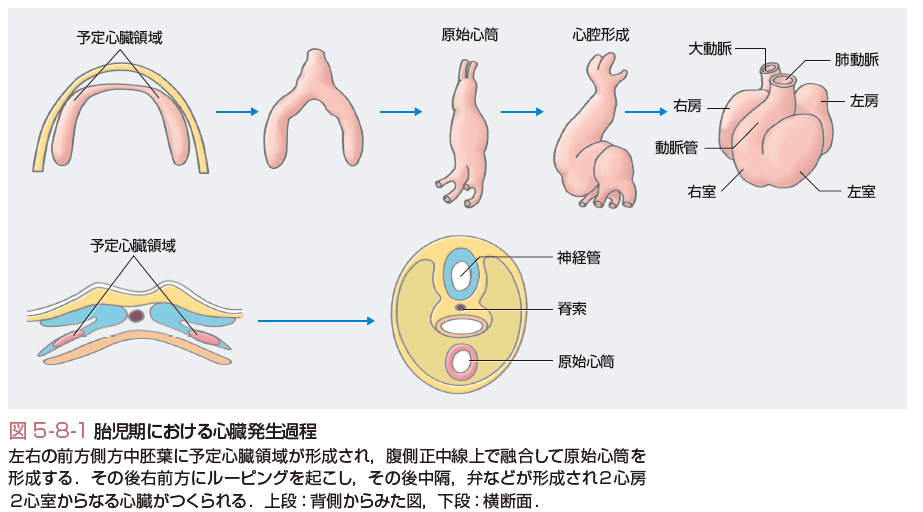
発生初期に前方側方中胚葉の予定心臓領域が正中線上で融合し,直線上の心筒(原始心筒)が形成される.さらに直線上の心筒は右側前方に向かって屈曲(ルーピング)し,心房は頭側に移動して心室の背側に位置するようになる.その後中隔,弁などが形成されて2心房2心室からなる心臓が形成される(図5-8-1).
先天性心疾患の頻度
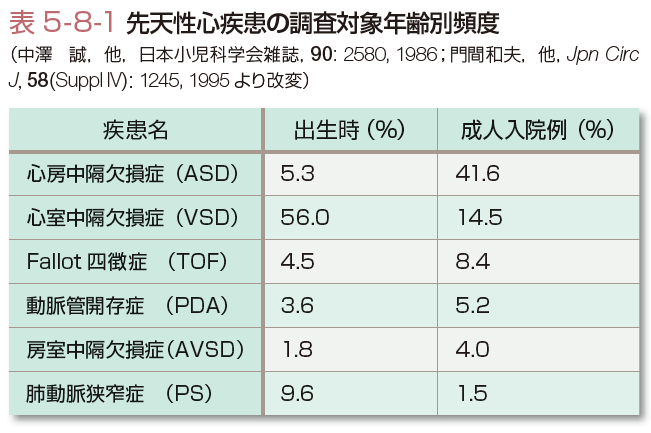
先天性心疾患は新生児の約1%にみられる先天異常である.出生時および成人入院例におけるおもな疾患とその頻度を示す(表5-8-1).出生時には心室性中隔欠損症が50%以上を占めるが,小児期までに自然閉鎖するものが多く,成人期では心房中隔欠損症の占める割合が相対的に増加する.
先天性心疾患の成因
先天性心疾患の病因としては,催奇形性因子・環境因子(薬物,風疹,母体の糖尿病,アルコール依存など)によるものが2%,染色体異常・遺伝子異常などの遺伝性要因が13%,その他特定はされていないが多因子遺伝と思われるものが85%とされる.すなわち先天性心疾患のほとんどは何らかの遺伝的要因の関与があることになる.ただ実際には単一遺伝子疾患であっても,胎内および出生後の環境因子や,ある遺伝子異常の表現型をさらに修飾する別の遺伝子(modifier gene)によって表現型が左右されるなど,遺伝子型と表現型は1対1で対応しない場合も多い.
先天性心疾患の分子機構
先天性心疾患の原因遺伝子の多くは現時点では特定されていないが,染色体異常あるいは遺伝子異常を伴う先天性心疾患の解析から,先天性心疾患発症の分子機構が徐々に明らかにされつつある.
1)22q11.2欠失症候群とTBX1:
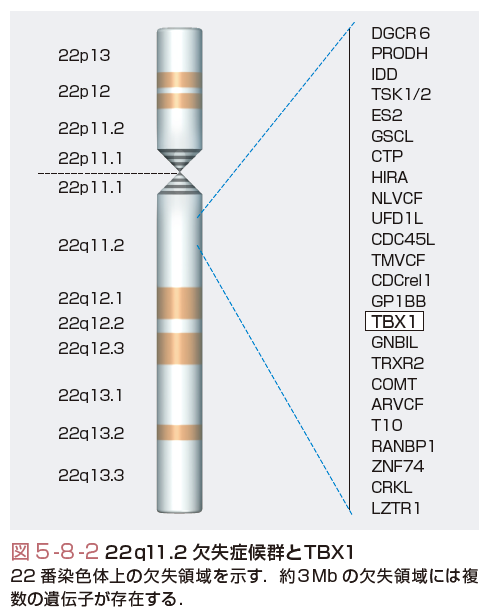
DiGeorge症候群,軟口蓋心臓顔貌症候群(velo-cardio-facial syndrome),円錐動脈幹異常顔貌症候群(conotruncal anomaly face syndrome)は一部重複した表現型を呈し,共通した染色体異常として2本の22番染色体のうち片方の長腕11.2の部分欠失を有することから,まとめて22q11.2欠失症候群とよばれる.22q11.2欠失症候群の臨床徴候はきわめて多彩で,特徴的顔貌(ほぼ100%),先天性心疾患(75%),免疫不全(胸腺形成不全による:2%),口蓋裂(9%),低カルシウム血症(副甲状腺形成不全による:47%),精神発達遅滞(50%)などを含む200以上の症状を呈する.22q11.2欠失症候群にみられる先天性心疾患としては,Fallot四徴症,大動脈弓離断症,心室中隔欠損症,総動脈幹症などが多くみられる.90%以上の症例においては共通する約3Mb(300万塩基対)の領域が欠失しており,この領域には30以上の遺伝子が存在するが,そのなかで特にTBX1が注目されている(図5-8-2).TBX1はT-boxとよばれるDNA結合領域をもつ転写因子ファミリー(T-boxファミリー)に属する転写調節因子で,二量体を形成して標的遺伝子の転写調節領域に結合し,その転写活性を制御する.Tbx1ヘテロ欠損マウスでは10〜50%に大動脈弓の異常がみられ,Tbx1ホモ欠損マウスでは100%に流出路異常,胸腺・副甲状腺形成不全,口蓋裂など22q11.2欠失症候群の表現型がみられた.また,ヒトにおいても,22q11.2欠失症候群の臨床症状を呈するにもかかわらず22q11.2に欠失を認めない家系においてTBX1の遺伝子変異が同定され,TBX1が22q11.2欠失症候群の主要な原因遺伝子であるものと考えられる.
2)Holt-Oram症候群とTBX5:
Holt-Oram症候群は上肢の奇形と先天性心疾患を呈する疾患で,先天性心疾患としては心房中隔欠損症が約半数を占め,そのほかに心室中隔欠損症や房室伝導障害などがみられる.1997年にHolt-Oram症候群の原因遺伝子としてTBX5が同定された.TBX5はTBX1と同様にT-boxファミリーに属する転写調節因子で,TBX5ヘテロ欠損マウスでは心房中隔欠損,心室中隔欠損,房室伝導障害,上肢骨格形成不全などヒトのHolt-Oram症候群ときわめて類似した表現型がみられる.したがってヒトのHolt-Oram症候群の機序としては,TBX5のhaplo-insufficiency(1対の相同染色体の一方の遺伝子の不活性化により表現型があらわれる)が考えやすい.実際これまでに複数のTBX5遺伝子変異が報告されているが,多くは機能喪失型の遺伝子変異であると考えられている.
3)家族性先天性心疾患とNKX2.5/CSX,GATA4:
NKX2.5/CSXとGATA4はいずれも心臓発生を制御する転写因子であり,DNA結合領域として前者はホメオドメイン,後者はZnフィンガードメインを有する.1998年になってNKX2.5/CSXが房室ブロックを伴う家族性心房中隔欠損症の原因遺伝子として同定され,その後の解析からNKX2.5/CSXの遺伝子変異により心房中隔欠損症だけでなく,心室中隔欠損症,Fallot四徴症,三尖弁異常などが起こることが明らかにされた.また,2003年にはGATA4が心房中隔欠損症,心室中隔欠損症,肺動脈狭窄症の原因遺伝子として同定された.TBX5,NKX2.5/CSX,GATA4の遺伝子変異では心房もしくは心室中隔欠損が多くみられ,これら3者の遺伝子変異の表現型にはかなりの共通性が認められる.これまでTBX5,NKX2.5/CSX,GATA4は互いに蛋白-蛋白間相互作用があり,協調的に標的遺伝子の転写を活性化することが知られていたが,家族性先天性心疾患でみられる遺伝子変異によりこれらの転写因子間の相互作用が失われる場合があることが明らかになった.TBX5,NKX2.5/CSX,GATA4の遺伝子変異の表現型に共通性が認められるのは,1つの転写因子の遺伝子変異によってこれら3種類すべての転写因子の標的遺伝子の発現が同時に影響を受けることがその理由の1つであると思われる.[塩島一朗]
出典 内科学 第10版内科学 第10版について 情報
Sponsored by ![]()