デジタル大辞泉
「重症筋無力症」の意味・読み・例文・類語
じゅうしょう‐きんむりょくしょう〔ヂユウシヤウキンムリヨクシヤウ〕【重症筋無力症】
運動神経からの興奮が筋肉にうまく伝わらず、脱力状態となる病気。初め眼筋に障害が現れ、進行すると四肢に広がり、呼吸筋が麻痺することもある。神経と筋との接合に自己免疫による異常があるために起こる。指定難病の一つ。
出典 小学館デジタル大辞泉について 情報 | 凡例
Sponsored by 
じゅうしょうきんむりょく‐しょうヂュウシャウシャウ【重症筋無力症】
- 〘 名詞 〙 運動を繰り返すことによって筋力が次第に低下し、休息によって回復するのを特徴とする疾患。全身の筋を冒すが、眼筋だけに起こる型もある。神経筋接合部におけるアセチルコリンの分泌異常に起因すると考えられている。若い成人女性に多い。厚生労働省特定疾患の一つ。MG。
出典 精選版 日本国語大辞典精選版 日本国語大辞典について 情報 | 凡例
Sponsored by
重症筋無力症(神経筋接合部疾患:重症筋無力症とLambert-Eaton筋無力症候群)
概念
重症筋無力症(myasthenia gravis:MG)は,神経筋接合部の後シナプス膜に対する自己免疫機序により刺激伝達が障害され,骨格筋の易疲労性・脱力をきたす自己抗体依存性神経疾患である.臨床的には,休息による回復,日内変動,および,寛解・増悪を特徴とする.後シナプス膜に存在するアセチルコリン受容体(acetylcholine receptor:AChR)や筋特異的受容体型チロシンキナーゼ(muscle-specific receptor tyrosine kinase:MuSK)などの機能蛋白質に対する自己抗体がおもな発症因子である(Farrugia,2010).
疫学
わが国の2006年MG臨床疫学調査では,患者概数15100人(男性5600人,女性9500人),有病率は人口10万人あたり11.8人と推測されている.男女比は1:2で,発症年齢の平均±標準偏差は,42.7±21.2歳であった.特に,50歳以上で発症するMG患者(late-onset MG)が増加していることが明らかになった.それに伴って,2010年日本神経治療学会から,late-onset MGの診断と治療の考え方を示す標準的治療指針が公表された.
病因・病態・病理
MGは自己抗体の種類によって,①AChR抗体陽性MG,②MuSK抗体陽性MG,そして,③前記の抗体が検出されないdouble seronegative MGに分類される.2011年,LDL受容体関連蛋白質4(LDL-receptor related protein 4:Lrp4)に対する自己抗体が報告され,AChR/MuSK抗体に次ぐ第3番目の病原性自己抗体として注目されている.わが国では,MG全体の約80%がAChR抗体陽性で,残りの5〜10%にMuSK/Lrp4抗体が検出される.自己抗体の作用機序は,IgG1が主体のAChR抗体は補体介在性に運動終板を破壊することによってMG症状を起こす.一方,抗MuSK抗体のサブクラスはIgG4が主体で補体介在性運動終板破壊がほとんどない神経筋接合部病理像が報告されている(図15-20-1,15-20-2).
分類
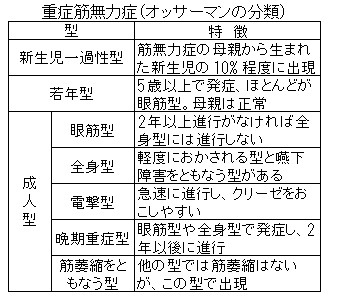
前記の自己抗体や臨床病型などによる分類がある.以前は1971年に発表されたOsserman分類がよく用いられていたが,2000年,米国重症筋無力症財団(Myasthenia Gravis Foundation of America:MGFA)より臨床研究を世界的に標準化するための臨床症状もしくは重症度に関するスケールとしてMGFA Clinical Classificationが提唱された.現在は本分類が一般的となっており,わが国の特定疾患臨床調査個人票でも用いられているが,どちらかというと分類ではなく重症度の指標として使用されている.具体的には,クラスⅠが眼筋型,クラスⅡ以降が全身型で,四肢筋,体幹筋の筋力低下が強ければaとなり,球症状が強ければbとなる.ⅡからⅤへ進むにつれて重症となり,Ⅴは気管内挿管された状態である.また,本症の母親から生まれる新生児の10~15%に胎盤を通過した抗体による一過性の本症がみられ,新生児一過性重症筋無力症といわれる.最近,MuSK抗体陽性MG患者からも新生児一過性重症筋無力症の報告が散見されている.
臨床症状
AChR抗体陽性MGの症状は,外眼筋が障害されやすい.物が二重に見えたり(複視),まぶたが無意識に下がったままの状態(眼瞼下垂)などの眼症状で発症し,全経過中ほとんどの症例にみられる.眼症状以外には,構音障害,嚥下障害,四肢麻痺が初期症状として多い.運動を反復することにより筋力低下をきたし休息によって改善する(易疲労性),朝より夕方に症状が出やすい(日内変動)などが特徴である.増悪因子として,ストレス,感染,月経,妊娠,分娩などがあげられ,これらを契機に嚥下困難や呼吸困難が急速に悪化する急性増悪(クリーゼ)に移行する. MuSK抗体陽性MGは,胸腺腫の合併はなく,筋萎縮を伴いやすく,嚥下障害が主体でクリーゼになりやすいという特徴をもつが,臨床レベルでAChR抗体かMuSK抗体かを鑑別することは困難である.
検査成績
1)塩化エドロホニウム(テンシロン,アンチレクス)試験:
コリンエステラーゼ阻害薬であるアンチレクス2~5 mgを静脈注射して反応をみる.臨床症状の著明な改善があるときを陽性とする.MuSK抗体陽性では,著効することは少なく,過敏反応がみられることがある.
2)電気生理学的検査:
顔面や四肢の筋を支配する末梢神経を反復刺激して,誘発筋活動電位を記録するHarvey-Masland試験では,低頻度刺激(1~5 Hz)でwaning 現象(初発刺激による振幅はほぼ正常で以後数発の刺激で振幅が10%以上減衰する)がみられる.また単一筋線維筋電図では,jitter(発射間隔の変動)の異常やブロッキング現象がみられる.
3)自己抗体測定:
AChR受容体は,MG全体の80%以上で陽性となる.ただし眼筋型では陽性率は約50%と低い.AChR抗体陰性患者のうち30~60%程度でMuSK抗体が陽性となる.AChR抗体価とMG症状の相関は,個々の症例で経時的にみると抗体価と疾患の重症度が相関する例があるが,患者を集団でみると抗体価と重症度は相関しない.一方,MuSK抗体価では抗体価と重症度は相関すると報告されている.
4)その他:
AChR抗体陽性MGでは,胸腺腫や胸腺過形成が存在することが多いため,胸部CT/MRI画像検査 などで評価する.
診断
上記の臨床症状,神経学的診察,および,検査所見に基づいて診断する.特に,病原性のあるAChR/MuSK抗体が陽性となれば確定と考えてよい.
鑑別診断
眼症状,球麻痺,および,四肢・体幹の筋力低下を呈する多くの疾患を鑑別する必要がある.眼症状に着目すれば,眼筋麻痺を呈する多くの疾患,動脈瘤(IC-PC)や糖尿病による動眼神経麻痺,Fisher 症候群,Tolosa-Hunt 症候群,ミトコンドリア脳筋症,眼・咽頭型ジストロフィー,Meige症候群,開眼失行などが鑑別の対象となる.球麻痺では,筋萎縮性側索硬化症,多発性硬化症,脳幹脳炎など,四肢筋力低下については,Guillain-Barré症候群,多発筋炎,筋ジストロフィー,そして,Lambert-Eaton 筋無力症候群などの鑑別が必要である.
合併症
胸腺腫は50歳をピークに20~30%にみられる.Basedow病などの甲状腺疾患が数%から十数%に合併する.それ以外に,赤芽球癆,全身性エリテマトーデス,関節リウマチなどの自己免疫疾患を合併する.
治療
コリンエステラーゼ阻害薬,胸腺摘除術(胸摘),ステロイド薬,免疫抑制薬(タクロリムス,シクロスポリン),血漿交換,そして,大量免疫グロブリン静注療法(2010年認可)などが保険適応として用いられている.胸腺腫合併例では,胸摘を行う.一方,胸腺腫非合併では,年齢,罹病期間,病型,重症度,胸腺画像,自己抗体の種類,そして合併症によって個々の症例で十分に検討されなければならない.以前には,全身型では胸摘を第一選択すべきとされてきたが,これに関しては有用なエビデンスがないことが判明し,現在,そのエビデンスを得るために治験,MGTX研究が進行中である.現状では,高齢発症MGでは胸腺異常の頻度は低く,胸腺摘除に関しては慎重であるべきである.眼筋型MGでは,その一部に自然寛解もあり,胸摘の施行は少なくとも発症初期には行うべきではない.半年から1年間はコリンエステラーゼ阻害薬や副腎皮質ホルモンで内科的に治療し,眼症状の再燃・難治例や全身型へ移行した例を中心に胸摘の適応を検討する.また,MuSK抗体陽性MGでは胸摘の有効性は低く,第一選択にはならない.治療法の詳細については神経治療学会のホームページに「MG治療のガイドライン」と「標準的神経治療:高齢発症MG」としてまとめられている. MGの臨床で一番問題になるのは,急激な筋力低下をきたし呼吸困難に陥ることがありクリーゼとよばれる.クリーゼには筋無力性とコリン作動性の2種類があり,テンシロン試験で改善すれば筋無力性と診断できるが,実際には区別が困難な場合が多い.よって,コリンエステラーゼ阻害薬はただちに中止し,気道を確保し呼吸管理を行うことが重要である.また,長期にステロイドを使用する場合が多く,骨粗鬆症予防や感染予防薬の投与を併用する.MGには,使用禁忌薬剤(ベンゾジアゼピン系薬,アミノ配糖体系抗菌薬,ダントロレンナトリウム,d-ペニシラミン,インターフェロン-αなど)が多数あるため注意を要する.
予後
MGの自然経過は明らかではないが,1965年以前の症例の検討によるとコリンエステラーゼ阻害薬のみの治療では約1/4の症例がMGのため発症3年以内に死亡している.その後,ステロイドと免疫抑制薬の導入により,MG患者の生命予後は劇的に改善した.ただし,わが国のMGクリーゼの頻度は,16.0%(1973年),14.8%(1987年),10.9%(2002年),13.3%(2006年)と報告され,まったく減少していない.今後は,このような重症かつ難治性MGにおける治療法の開発が期待される.[本村政勝]
■文献
Farrugia ME, Vincent A : Autoimmune mediated neuromuscular junction defects. Curr Opin Neurol, 23: 489-495, 2010.
本村政勝,白石裕一:抗MuSK抗体陽性重症筋無力症.In Annual Review 神経(鈴木則宏,祖父江 元,他編)pp328-336,中外医学社,東京,2011.
本村政勝,福田 卓:【神経筋接合部 基礎から臨床まで】 Lambert-Eaton筋無力症候群.Brain Nerve, 63: 745-754, 2011.
Shiraishi H, Motomura M, et al: Acetylcholine receptors loss and postsynaptic damage in MuSK antibody-positive myasthenia gravis. Ann Neurol, 57: 289–293, 2005.
出典 内科学 第10版内科学 第10版について 情報
Sponsored by
家庭医学館
「重症筋無力症」の解説
じゅうしょうきんむりょくしょう【重症筋無力症 Myasthenia Gravis】
◎著しく疲れやすくなる
[どんな病気か]
著しい易疲労性(いひろうせい)(疲れやすさ)と日内変動(にちないへんどう)(病状が1日のなかで変わる)が症状の中核となる病気です。厚労省の特定疾患(とくていしっかん)(難病)に指定され、医療費は公費からの補助が受けられます。
患者さんは、若い成人では女性が、中年以降では男性が多くなっています。また、子どもでは、眼筋型(がんきんがた)というタイプが多くみられます。
病状や症状によって、いろいろなタイプに分類されています(表「重症筋無力症(オッサーマンの分類)」)。
[原因]
自己免疫疾患(じこめんえきしっかん)の1つです。自己免疫疾患とは、自分のからだの組織を敵とみなす抗体(こうたい)ができ、抗体がその組織を攻撃して破壊してしまう疾患群をさします。
重症筋無力症の場合は、神経筋接合部(運動終板(うんどうしゅうばん)=運動神経と筋肉のつなぎ目)に存在するアセチルコリン受容体(じゅようたい)(レセプター=物質を取り入れる細胞の取り入れ口)に対する抗体ができ、受容体が破壊されます。すると、神経の命令を細胞に伝える伝達物質であるアセチルコリンが、筋肉細胞の中に入れなくなります。そのために神経の命令が筋肉に伝わらず、筋肉を動かしづらくなって、疲れやすくなるのです。
[症状]
物が二重に見えたり(複視(ふくし))、まぶたが下がって(眼瞼下垂(がんけんかすい))、眠そうな顔つきになります。
からだを動かすことはもちろん、物をかむだけでも疲れるという著しい疲れやすさ(易疲労性)が特徴です。また、この症状は朝は軽く、夕方になるとひどくなります(日内変動)。
ときに、生命に危険がおよぶほどに急激に悪化する、クリーゼという状態になることもあります。
[検査と診断]
採血をして、血液中に含まれるアセチルコリン受容体に対する抗体の値を測定します。なかには異常値を示さないケースもあります。
末梢運動神経を連続的に刺激する筋電図検査も有効です。不要になったアセチルコリンは、(アセチル)コリンエステラーゼという酵素(こうそ)によって分解されていますが、分解されなければ、筋肉への伝達作用は増長されると考えられます。
そこで、この酵素のはたらきを一時的に抑えてようすをみるテンシロンテストを行ないます。
この検査では、酵素のはたらきを抑える、作用時間の短い、抗コリンエステラーゼ剤(テンシロン)を静脈に注射します。重症筋無力症の場合、症状が劇的に改善します。
症状が急激に悪化するクリーゼの原因としては、筋無力症自体の悪化や、抗コリンエステラーゼ剤の使いすぎが考えられます。この場合も、テンシロンテストを行なえば、どちらか判定できます。
重症筋無力症は、胸腺腫瘍(きょうせんしゅよう)をともなうことも多いので、CTなどで胸を撮影し、その有無を調べます。
◎アセチルコリンの作用増強が決め手
[治療]
神経末端から放出されるアセチルコリンを神経筋接合部で分解してしまうコリンエステラーゼの作用を抑え、アセチルコリンのはたらきを増強させるのが治療の基本です。そのため、抗コリンエステラーゼ剤を服用しますが、テンシロンよりも作用時間の長い薬を使用します。
複視や眼瞼下垂以外の症状が出現するようであれば、胸腺を摘出する手術が考慮されます。
手術後、一時的に症状が悪化することがありますが、長期的には手術の有効性が明らかになっています。
異常な免疫を抑えるために副腎皮質(ふくじんひしつ)ホルモン薬や免疫抑制薬(めんえきよくせいやく)のアザチオプリンも使用します。
血液中のアセチルコリン受容体に対する抗体を取り除く血漿交換療法(けっしょうこうかんりょうほう)(いったん血液を体外へ導き出し、目的とする物質を取り除いてから、また体内へもどす治療)も有効です。
出典 小学館家庭医学館について 情報
Sponsored by
重症筋無力症
じゅうしょうきんむりょくしょう
myasthenia gravis
代表的な難病(厚生労働省特定疾患)の一つにあげられており、しばしば頭文字からMGと略称されている。神経‐筋接合部の障害によって四肢の筋の運動を繰り返すと脱力が生じ、休息によってふたたび筋力が戻ってくる特異な症状を示すのが特徴である。原因は不明であるが、神経から興奮を伝える物質アセチルコリンの受容体のタンパク質に対する抗体が血液中に現れ、これが筋肉にある受容体と結合して受容体を壊してしまうために、神経からの興奮が手足の筋肉に十分伝わらず、脱力をおこすことが明らかとなった。血清中にあるこれらの抗体値を測定し、高い場合には血漿(けっしょう)交換をすると症状が速やかに改善することも明らかとなり、現在、自己免疫疾患の代表と考えられている。
MGは新生児から老人まで、どの年齢層にもおこるが、新生児では母親がMGである場合、一過性に症状の発現をみるのみである。日本では5歳以下と30歳代に多く、女性が男性の約2倍である。症状は眼瞼(がんけん)下垂か眼筋麻痺(まひ)で始まることが多く、進行すると嚥下(えんげ)、呼吸、四肢の筋力低下を生じ、ときに急性増悪(クリーゼ)して呼吸麻痺をおこす。大部分の患者は胸腺(きょうせん)の肥大をもち、一部は胸腺腫(しゅ)を合併している。筋の脱力は運動により増悪し、安静により回復するが、日内変動するのが特有で、抗コリンエステラーゼ剤を投与すると、ただちに一過性の症状改善がみられる。これは診断法として用いられる。治療には、対症療法として抗コリンエステラーゼ剤の投与が行われ、一過性の筋力維持に使われているが、根本療法として胸腺摘出術、免疫抑制剤投与、血漿交換法などにより、約7割の患者は完全寛解をみるようになった。
[里吉営二郎]
出典 小学館 日本大百科全書(ニッポニカ)日本大百科全書(ニッポニカ)について 情報 | 凡例
Sponsored by
百科事典マイペディア
「重症筋無力症」の意味・わかりやすい解説
重症筋無力症【じゅうしょうきんむりょくしょう】
筋無力症とも。筋肉の神経障害により筋力が低下し筋肉が疲労しやすくなる疾患。20代の女性に多く,感染症,精神的興奮は誘因となる。まず顔面,動眼,咽頭(いんとう),喉頭(こうとう),呼吸筋などが冒され,眼瞼(がんけん)下垂,複視等の症状を示し,鼻声となり,誤嚥(ごえん)を起こしやすく,進行すれば全身の筋の脱力を生ずる。筋萎縮(いしゅく),知覚障害は伴わない。代表的な自己免疫疾患である。治療には抗コリンエステラーゼ剤が有効で,この副作用を防ぐためにアトロピンを併用する。全身型では胸腺摘出術を行うこともある。副腎皮質ホルモン,血漿(けっしょう)交換療法もときに行われる。自然緩快,自然治癒(ちゆ)もみられる。→筋萎縮症
→関連項目ガランタミン|睡眠時無呼吸症候群|鉄の肺|難病
出典 株式会社平凡社百科事典マイペディアについて 情報
Sponsored by
重症筋無力症 (じゅうしょうきんむりょくしょう)
myasthenia gravis
若い成人女性に多く,随意筋が疲れやすく,動作を反復持続することが困難となり,ついには麻痺を呈する疾患。疲労や麻痺は休息によって回復する。難病の一つ。とくに眼瞼下垂,眼球運動の障害,構音障害,嚥下障害を起こしやすい。症状に悪化と軽快の波があり,1日のうちでも,朝は障害が軽く,夕方に向かうにつれて症状が強くなる。筋電図では,徐々に活動電位が低下する減衰現象がみられる。症状は抗コリンエステラーゼ剤の使用によって軽快することから,この疾患の診断にも利用される。神経筋接合部での興奮の伝達が障害されており,筋肉にあるアセチルコリン受容体に対する抗体価が高くなることや,胸腺の過形成,胸腺腫あるいは他の自己免疫疾患が合併することなどから,自己免疫疾患であると考えられている。治療としては,抗コリンエステラーゼ剤や副腎皮質ホルモン剤の投与のほか胸腺摘出術が行われるが,免疫抑制剤を用いたり,血漿交換療法なども行われる。
執筆者:水沢 英洋
出典 株式会社平凡社「改訂新版 世界大百科事典」改訂新版 世界大百科事典について 情報
Sponsored by
重症筋無力症
じゅうしょうきんむりょくしょう
myasthenia gravis; MG
軽い運動でも骨格筋が容易に疲労し,休息で回復することを特徴とする疾患。自己免疫疾患と考えられている。原因としては,神経筋接合部の代謝異常説が最も有力である。すなわち,筋終板におけるアセチルコリンの絶対量の減少,アセチルコリンを分解する酵素の過剰生成,アセチルコリンに対する感受性の低下などの諸説がある。病型は,新生児一過性,若年性,成人型に分けられる。病状は朝起床時には比較的よいが,午後,特に夕方になると筋肉の脱力が強くなる。眼瞼下垂,眼球運動障害とそれによる複視,口唇筋や頬筋の麻痺,嚥下,咀しゃく,言語の障害が現れる。四肢筋では特に下肢筋の脱力が強い。進行すると球麻痺,呼吸筋麻痺を起すため,予後が不良となる。治療には,抗コリンエステラーゼ剤 (テンシロン,ワゴスチグミン,マイテラーゼ,メスチノン) が用いられ,副腎皮質ホルモンも有効である。胸腺腫を合併することが多く,その場合には,外科的に摘出する。
出典 ブリタニカ国際大百科事典 小項目事典ブリタニカ国際大百科事典 小項目事典について 情報
Sponsored by
重症筋無力症
神経筋接合部で刺激伝導に障害があり,筋肉が疲労しやすくなったり脱力したりする疾病.
出典 朝倉書店栄養・生化学辞典について 情報
Sponsored by

{kind=link}