デジタル大辞泉
「性分化疾患」の意味・読み・例文・類語
せいぶんか‐しっかん〔セイブンクワシツクワン〕【性分化疾患】
出典 小学館デジタル大辞泉について 情報 | 凡例
Sponsored by 
性分化疾患(内分泌系の疾患)
(3)性分化疾患(disorder of sex development:DSD)
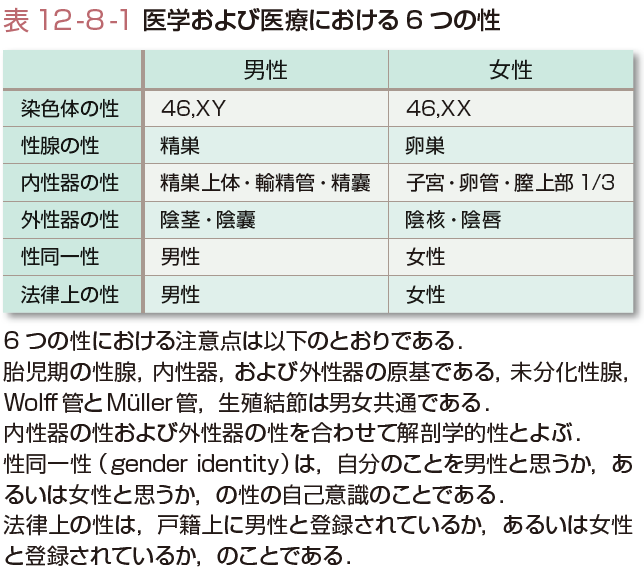
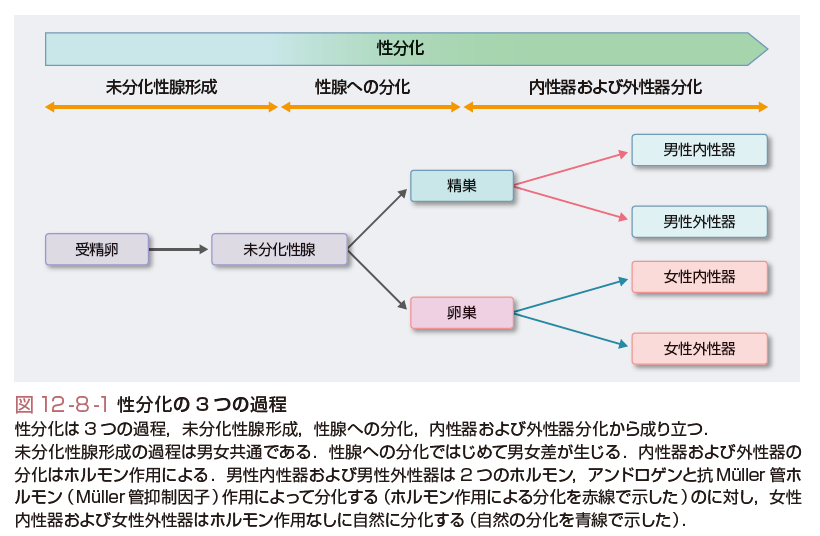
性分化疾患は上記の性分化過程の障害による疾患であり,染色体,性腺,または解剖学的性(内性器の性および外性器の性)が非定型的である先天的状態と定義される.したがって,性分化疾患では医学および医療で考える6つの性のうち,染色体の性,性腺の性,解剖学的性(内性器の性および外性器の性)に焦点を当てている.性分化疾患に該当する用語として,過去にインターセックス,半陰陽,性分化異常症などが用いられていたが,これらは患者と家族にとって蔑視的な意味が潜むと感じられることから,現在では性分化疾患という用語に統一された.
分類
性分化疾患は表12-8-2のように分類される.
疫学
諸外国の文献上のデータから,約4500人に1人と推定されているが,わが国における正確な疫学の成績はない.
性分化疾患各論
以下,性分化疾患のうち比較的頻度の高い6疾患について概説する.なお,46,XY DSDであるSTAR異常症,46,XX DSDである21-ヒドロキシラーゼ欠損症,11β-ヒドロキシラーゼ欠損症,46,XY DSD かつ46,XX DSDであるP450オキシドレダクターゼ欠損症については副腎ステロイド合成異常症を参照のこと.【⇨12-6-10)】
a.Turner症候群
定義・概念
Turner症候群は1本のX染色体すべてあるいは短腕の一部を欠き,低身長,Turner身体徴候(後述),原発性卵巣機能低下症などの特徴的な臨床症状を呈する女性であり,典型的な染色体核型は45,Xである.Turner症候群は性染色体異常に伴う性分化疾患の代表である(表12-8-2,12-8-3).
原因・病因
両親の配偶子(精子あるいは卵子)形成の不分離により,45,Xを生じる.
疫学
女児出生1/2000~1/2500.Turner症候群の胎児の約95%は自然流産するといわれている.
病理
Turner症候群の病理組織学的特徴は2点に集約される.第一の特徴はリンパ管低形成である.軟部組織におけるリンパ管低形成は,リンパ管内あるいは組織内のリンパ液うっ滞をきたし,翼状頸,手背および足背の浮腫などのTurner身体徴候(後述)をきたす.第二の特徴は索状性腺である.胎生初期の45,X胎児の卵巣は組織学的に正常であるが,次第に異形成に転じる.すなわち,卵祖細胞は正常であるが,第一減数分裂を開始した卵母細胞はアポトーシスにより急激に細胞死する.卵母細胞非存在下には卵胞は形成されず,結果的に卵胞形成に伴う顆粒膜細胞および莢膜細胞の分化は起こらない.最終的には索状性腺となる.
病態生理
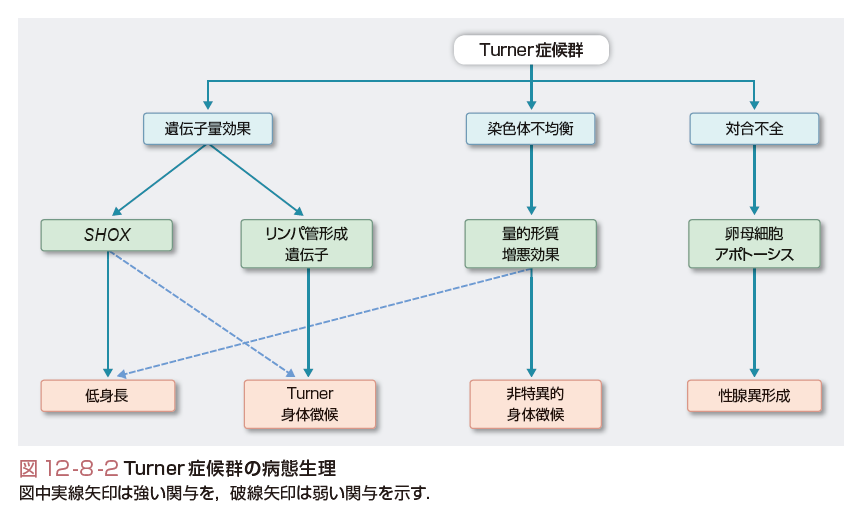
Turner症候群の病態生理は遺伝子量効果,染色体不均衡,染色体対合不全の3つの組み合わせで説明される(図12-8-2).①-1 X染色体偽常染色体領域に存在するSHOX遺伝子の欠失(遺伝子量効果)が低身長の主因である.SHOX遺伝子はX染色体およびY染色体短腕偽常染色体領域に存在し,不活化をまぬがれる遺伝子である.SHOX蛋白質はホルモン非依存性に骨成長を促進する.46,XX個体において,SHOX遺伝子は2コピー発現するが,45,X個体では1コピーしか発現しない.①-2 X染色体短腕に存在が推定されている“リンパ管形成遺伝子”の欠失(遺伝子量効果)がTurner身体徴候の主因である.“リンパ管形成遺伝子”はクローニングされていないが,不活化をまぬがれる遺伝子と考えられる.45,X個体では“リンパ管形成遺伝子”が1コピーしか発現しないことにより,リンパ管低形成をきたすと考えられる.②X染色体異常という染色体不均衡が量的形質増悪効果を介する非特異的身体徴候の主因である.染色体不均衡は一部低身長にも関与する.③減数分裂の際の染色体対合不全が卵母細胞のアポトーシス(上述)を介する索状性腺の主因である.
臨床症状
低身長,Turner身体徴候,原発性卵巣機能低下症を主症状とする.低身長はTurner症候群の95~100%に認め,日本人無治療Turner症候群の成人身長は平均139 cmである.Turner身体徴候の代表は翼状頸(図12-8-3),手背あるいは足背のリンパ性浮腫(図12-8-4),外反肘などである.Turner症候群に原発性卵巣機能低下症は必発である.典型的には重症の原発性卵巣機能低下症であり,80%の患者は二次性徴を欠如する.20%の患者において認められる二次性徴も不完全であり,99%以上の患者は不妊である.
検査成績
血清ゴナドトロピン高値を示す.
診断
特徴的な臨床症状からTurner症候群を疑い,染色体検査で診断を確定する.
鑑別診断
Noonan症候群:
身体所見(低身長およびTurner身体徴候)は似ているが,染色体核型は46,XYまたは46,XXである.
合併症
1)循環器:
大動脈縮窄症,大動脈二尖弁を代表とする先天性疾患,大動脈拡張,高血圧を代表とする後天性疾患ともに多い.
2)腎泌尿器:
馬蹄腎などの先天性疾患を認めることがある.
3)内分泌代謝疾患:
年齢が進むにつれ,肥満,糖尿病,慢性甲状腺炎の合併が増える.
4)その他:
肝機能障害,骨粗鬆症,難聴などを認めることがある.知能は正常であるが,動作性IQが低下するといわれている.
治療
低身長に対し成長ホルモン投与が行われ,成人身長は147 cm前後に改善する.
原発性卵巣機能低下症に対し,女性ホルモン補充療法が行われる.
b.Klinefelter症候群
定義・概念
Klinefelter症候群は染色体核型47,XXYで特徴づけられる疾患であり,性染色体異常に伴う性分化疾患の代表である(表12-8-2,12-8-3).
分類
47,XXY以外の過剰なX染色体に起因する男性に発症する疾患群(48,XXXY,49,XXXXYなど)すべてをKlinefelter症候群の亜型と考える分類もある.
原因・病因
両親の配偶子(精子あるいは卵子)形成の不分離により,47,XXYを生じる.
疫学
男児出生1/500~1/1000.
病理
Klinefelter症候群の精巣組織の特徴は,精細管の線維化および硝子化,精子形成の欠如である.
病態生理
Klinefelter症候群の病態生理もTurner症候群と同様に遺伝子量効果,染色体不均衡,染色体対合不全の3つの組み合わせで考えると理解しやすい.①X染色体偽常染色体領域に存在するSHOX遺伝子の過剰(遺伝子量効果)が高身長の主因である.SHOX遺伝子は47,XXY個体では3コピー発現する.②臨床症状への寄与は必ずしも明確ではないが,X染色体異常という染色体不均衡が存在する.③減数分裂の際の染色体対合不全が無精子症の主因と考えられている.
臨床症状
1)自覚症状:
高身長,女性化乳房,長い手足,やせ,不妊(無精子症による)などを認める.
2)他覚症状:
小精巣は必発の所見である.
検査成績
血中ゴナドトロピン(特にFSH)高値を示す.
診断
高身長,女性化乳房あるいは不妊からKlinefelter症候群を疑い,染色体検査で診断を確定する.
鑑別診断
Klinefelter症候群以外の男性不妊をきたす疾患.
合併症
平均IQは85~90といわれている.乳癌発症リスクは健常男性の20倍である.
治療
近年,Klinefelter症候群の不妊(無精子症)に対し,顕微鏡下精巣内精子採取法(microdisection-testicular sperm extraction:MD-TESE)と顕微授精(intracytoplasmic sperm injection:ICSI)を組み合わせた挙児獲得の報告が散見される.顕微鏡下精巣内精子採取法とは,顕微鏡を用いて精巣中をくまなく観察し,精子がいる可能性のある太い精細管から精子採取を試みる手術である.顕微鏡下精巣内精子採取法によりKlinefelter症候群の約半数で精子採取が可能であったとの報告もある.
c.SF1異常症
定義・概念
SF1異常症は46,XY DSDのうち,部分型性腺異形成の代表的疾患である(表12-8-2,12-8-3).なお,46,XX個体におけるSF1異常症は必ず性腺異形成をきたすわけではない.
原因・病因
SF1遺伝子の異常に起因する.多くは1アレル性変異で発症する.
病理
SF1異常症の典型的な性腺組織は精巣異形成(dysgenetic testes)である.
病態生理
未分化性腺形成の障害のため,46,XY個体において精巣への分化は不十分である.結果的に胎児期の精巣からの2つのホルモン分泌が低下する.
臨床症状
尿道下裂,矮小陰茎,などの外性器異常をきたす.生下時に法律上の性の決定に迷うこともまれではない.
検査成績
46,XY個体においてしばしばhCG負荷試験に対するテストステロンの反応性は低下している.
診断
臨床症状および検査成績から本症を疑うことは必ずしも容易ではない.SF1遺伝子解析により診断を確定する.
鑑別診断
SF1異常症以外の46,XY個体における部分型性腺異形成.
治療
法律上の性に合わせて,必要に応じて性腺摘出術,外性器形成術を行う.性腺を摘出した法律上の女性に対して女性ホルモン補充療法を行う.
d.5α還元酵素欠損症
定義・概念
5α還元酵素欠損症は精巣が分泌するアンドロゲンであるテストステロンを生体内で最も強力なアンドロゲンであるジヒドロテストステロンに変換する酵素である5α還元酵素の欠損による.46,XY DSDのうち,アンドロゲン生合成障害の代表的な疾患である(表12-8-2,12-8-3).
分類
ジヒドロテストステロン産生低下の程度が重症なタイプと軽症なタイプに分けられる.
原因・病因
5α還元酵素を規定するSRD5A2遺伝子異常による常染色体劣性遺伝病である.
病態生理
ジヒドロテストステロン産生低下のため,外性器が完全に男性化しない.
臨床症状
症例により臨床症状の差異を認める.ジヒドロテストステロン産生低下の程度が重症なタイプの外性器は陰核肥大および陰唇様にみえ,生下時に法律上の性を女性と決定されることもある(図12-8-5).ジヒドロテストステロン産生低下の程度が軽症なタイプは矮小陰茎のみを呈する(図12-8-6).
検査成績
血中テストステロン/ジヒドロテストステロンの比は上昇する.
診断
血中テストステロン/ジヒドロテストステロンの比上昇から本症を疑い, SRD5A2遺伝子解析により診断を確定する.
鑑別診断
不完全型アンドロゲン不応症(後述).
治療
法律上の女性では通常性腺摘出術,外性器形成術を行い,女性ホルモン補充療法を行う.法律上の男性ではジヒドロテストステロン軟膏による治療を行う.
e.アンドロゲン不応症
定義・概念
アンドロゲン不応症はアンドロゲンに対する受容体レベルでの不応に起因する.46,XY性分化疾患のうち,アンドロゲンあるいは抗Müller管ホルモン合成障害・作用異常の代表的疾患である(表12-8-2,12-8-3).
分類
完全型アンドロゲン不応症と不完全型アンドロゲン不応症に分類される.完全型は外性器完全女性型を示し,生下時に逡巡なく法律上の性を女性と決定される.不完全型は外性器不完全女性型,不完全男性型,あるいは完全男性型を示す.外性器不完全女性型あるいは不完全男性型の際には生下時に法律上の性の決定に苦慮することもある.
原因・病因
46,XY個体におけるX染色体に存在するアンドロゲン受容体のヘミ接合性変異による.したがって本症はX連鎖劣性遺伝病である.
疫学
1/20000~1/64000.
病態生理
精巣から胎生期および思春期にアンドロゲンが正常に分泌されるが,アンドロゲン受容体レベルでさまざまな程度の不応性を有するため,外性器を含む男性化障害をきたす.
臨床症状
症例により臨床症状の差異を認める.
1)完全女性型外性器:
外性器は完全女性型を示す.子宮を欠如し,膣は盲端に終わる.陰毛および腋毛を欠如する.精巣は鼠径部あるいは腹腔内に存在する.思春期年齢に乳房の発達を認めるが,原発性無月経である.
2)男性化徴候を伴う女性型外性器:
陰核肥大,陰唇融合などの外性器男性化を認める(図12-8-7).精巣は陰唇内,鼠径部あるいは腹腔内に存在する.
3)不完全男性型外性器:
矮小陰茎,尿道下裂,二分陰囊などを認める(図12-8-8).精巣はしばしば停留精巣である.
4)完全男性型外性器:
外性器は完全男性型であるが,無精子症を有し男性不妊である.
検査成績
血中LH高値,テストステロンおよびジヒドロテストステロン正常,hCG負荷試験に対してテストステロンの反応性正常である.染色体核型は46,XYである.
診断
臨床症状および検査成績から本症を疑う.X連鎖劣性遺伝と矛盾しない家族歴を有する際には診断は容易である.家族歴を有さない際はアンドロゲン受容体遺伝子解析により診断を確定する.
鑑別診断
不完全型アンドロゲン不応症ではジヒドロステロン産生低下が重症であるタイプの5α還元酵素欠損症(前述).
合併症
鼠径ヘルニア(図12-8-7)(女児鼠径ヘルニアの1~2%は本症といわれている).
治療
法律上の性に合わせて,必要に応じて性腺摘出術,外性器形成術を行う.性腺を摘出した思春期年齢到達後女児は女性ホルモン補充療法を行う.
f.Müller管形成不全症
定義・概念
46,XX個体において子宮,卵管,膣上1/3が欠損する疾患である(表12-8-2,12-8-3).Mayer-Rokitansky-Kuster-Hauser症候群ともよばれる.
原因・病因
女性内性器原器であるMüller管の発生異常によると考えられる.Müller管形成不全症に男性化,片腎などを伴う疾患はWNT4遺伝子異常による.
疫学
1/5000女児出生.
臨床症状
二次性徴としての乳房発達は正常である.しかし,子宮欠損のために初経を認めず(原発性無月経),不妊である.
検査成績
内分泌学的な異常を認めない.染色体核型は46,XXである.
診断
初経を認めない46,XX女性において,卵巣機能は正常であること,かつ画像診断で子宮の欠損を証明すれば診断は確定する.
合併症
WNT4遺伝子の異常に起因するMüller管形成不全症では多毛などの男性化徴候,片腎などを合併する.[長谷川奉延]
■文献
Hughes IA, Houk C, et al: Consensus statement on management of intersex disorders. Arch Dis Child, 91: 554-563, 2006.
出典 内科学 第10版内科学 第10版について 情報
Sponsored by
性分化疾患
せいぶんかしっかん
differences of sex development
性染色体、性腺(せいせん)、内性器あるいは外性器のいずれかが大多数の男性や女性とは異なる、非定型的な先天的状態。したがって、性分化疾患は一つの疾患ではなく、60以上の疾患あるいは症候群の総称である。DSDと略称される。
2006年、専門家による国際会議で、統一された国際名称disorders of sex development(DSD)が発表された。それまでは半陰陽hermaphroditismという診断名が用いられていたが、患者のなかに半陰陽という言葉は蔑視(べっし)的な意味を含むと感じる人がいたためである。さらに、DSDの概念が提唱された2006年当初は、性分化異常症とよばれていたものが、DSDは異常ではなく多様性の一つである、あるいは個性であるとの考え方が普及し、近年では国際的にはdifferences of sex development(DSD)とされることが多い。日本では「性分化疾患」という名称が多く用いられている。
[長谷川奉延 2026年1月20日]
諸外国のデータから、DSDのうち男児か女児かの判別不明の非定型的外陰部を有する症例は、出生児のおよそ4500人に1人と推定されている。日本における信頼性の高い疫学研究はない。性染色体異常に伴うDSD(後述)のうち、男性にもっとも多いクラインフェルター症候群(代表的な染色体核型は47,XXY〈多くの健常男性の染色体核型は46,XY〉)は男性およそ650人に1人、女性にもっとも多いターナー症候群(代表的な染色体核型は45,X〈多くの健常女性の染色体核型は46,XX〉)は女性およそ2000人に1人である。DSDの家族歴、あるいは妊娠母体の胎盤機能不全はDSDの危険因子である。
[長谷川奉延 2026年1月20日]
性染色体の核型により、「性染色体異常に伴うDSD」「46,XY DSD」「46,XX DSD」の三つに分類される。
症状としては出生時の非定型的外陰部、法律上の性と合致する二次性徴の未出現あるいは進行不全、法律上の性と合致しない二次性徴の出現、不妊などを呈する。DSDのなかでも個々の疾患あるいは症候群により症状の差異は大きい。
[長谷川奉延 2026年1月20日]
診断は次の検査を組み合わせて行われる。
①染色体検査
②内分泌学的検査(血液中の性ホルモン〈テストステロン、エストラジオールなど〉の測定など)
③画像検査(内性器および性腺の形態を確認するための超音波検査、MRIなど)
④遺伝子検査
[長谷川奉延 2026年1月20日]
①非定型的外陰部を有する新生児の法律上の性の選択
新生児の法律上の性の決定権を有する親権者とともに、将来のジェンダー・アイデンティティ(性同一性)を予測したうえで法律上の性を選択する。ただし、非定型的外陰部を有する新生児の将来のジェンダー・アイデンティティを、新生児期の外陰部の形状のみから予測することはむずかしいことがわかっている。そのため、種々の検査などを通して慎重な検討がなされることが望ましい。戸籍法には出生後14日以内に出生届(性別・氏名)を提出と明記されているが、期限延長も可能である。
②外科的手術
選択した法律上の性により適合するように、外陰部形成術および不要な性腺摘出術を行う。
③薬物治療
選択した法律上の性により適合するように、二次性徴を誘導・維持するホルモン補充療法を行う。
④心理社会的な支援
選択した法律上の性に合致したジェンダー・アイデンティティを確立・維持できるように、家族とともに本人を支援する。本人にDSDを理解したうえで受容してもらえるように、段階的かつ繰り返しDSDについて説明する。
[長谷川奉延 2026年1月20日]
おのおのの疾患あるいは症候群により経過・予後は異なる。46,XY DSDで組織学的に未熟な性腺を有する患者は、性腺腫瘍(しゅよう)発症のリスクを有する。
[長谷川奉延 2026年1月20日]
出典 小学館 日本大百科全書(ニッポニカ)日本大百科全書(ニッポニカ)について 情報 | 凡例
Sponsored by