改訂新版 世界大百科事典 「有機化学反応」の意味・わかりやすい解説
有機化学反応 (ゆうきかがくはんのう)
organic chemical reaction
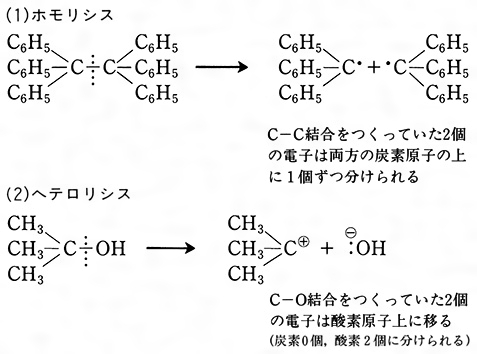
有機化学反応に限らず化学反応一般において,反応物が生成物に変換されるためには,反応物をつくる結合の開裂,それと同時に反応物をつくる結合の生成が起こる必要がある。この結合の開裂(および生成)には二つの様式がある。原子AとBが2個の電子を共有してつくる結合A:Bの開裂が対称的に起こり,AおよびBがそれぞれ1個の電子を得るとき,すなわち開裂が
A:B─→A・+・B ……(1)
のように進むとき,この様式を均等開裂homolytic cleavageという。生成したA・および・Bは(それが1個の原子からなる場合も含めて),不対電子を含めて奇数個の価電子をもち,ラジカルradical(フリーラジカル,遊離基ともいう)と呼ばれる。不対電子を炭素原子上にもつ炭素ラジカル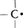 は7個の価電子をもち,一般にきわめて反応性が高く単離が困難である場合が多い。これに対して結合A:Bが非対称に開裂し,AまたはBのいずれかが結合電子対を得るとき,すなわち
は7個の価電子をもち,一般にきわめて反応性が高く単離が困難である場合が多い。これに対して結合A:Bが非対称に開裂し,AまたはBのいずれかが結合電子対を得るとき,すなわち
A:B─→A:⁻+B⁺またはA⁺+:B⁻ ……(2)
のような開裂を不均等開裂heterolytic cleavageという。生成物は正電荷または負電荷をもち,いずれもイオンである。A,Bにおいて正電荷が炭素原子上にあるものはカルボカチオン(カルボニウムイオンcarbonium ionとも呼ばれる)である。カルボカチオン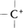 は6個の価電子をもつ電子不足性の分子種で反応性に富み,電子過剰性分子種と反応しやすい。これに対して炭素原子上に負電荷をもつカルバニオンは,電子過剰性分子種で他の原子または分子種と価電子を共有する必要がある。いずれにしても炭素原子がつくる結合の開裂によってラジカル,イオンのいずれかが生じ,これらが種々の試薬と反応して生成物となるのが有機化学反応の原型である。
は6個の価電子をもつ電子不足性の分子種で反応性に富み,電子過剰性分子種と反応しやすい。これに対して炭素原子上に負電荷をもつカルバニオンは,電子過剰性分子種で他の原子または分子種と価電子を共有する必要がある。いずれにしても炭素原子がつくる結合の開裂によってラジカル,イオンのいずれかが生じ,これらが種々の試薬と反応して生成物となるのが有機化学反応の原型である。
ラジカル反応とイオン反応
有機化学反応を結合の開裂様式で分類する代りに,中間に生じる活性分子種の種類によって反応を分類することもしばしば行われている。均等開裂をともなう反応をラジカル反応,不均等開裂をともなう反応をイオン反応と呼ぶのが一般的であるが,後者を極性反応polar reactionと呼ぶこともある。イオン反応は室温,暗所(光を特に必要としない),溶液中で起こることが多い。水やエチルアルコールなどの極性溶媒はイオンの生成に有利である。ラジカル反応は高温,太陽光下,気相で起こりやすい。
置換,付加,脱離,転位
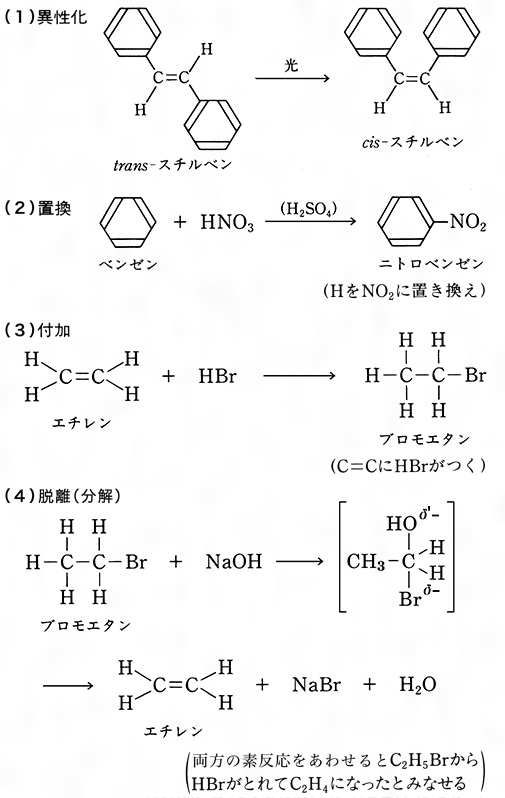
結合の開裂様式に基づく分類とは別に,反応物と生成物の構造の関係に基づく反応の分類も実用上きわめて便利である。置換substitution反応は,二つの反応物がその一部を交換する反応で,一般的には
A:B+X:Y─→A:X+B:Y ……(3)
で表される。ベンゼンのニトロ化などは典型的置換反応である。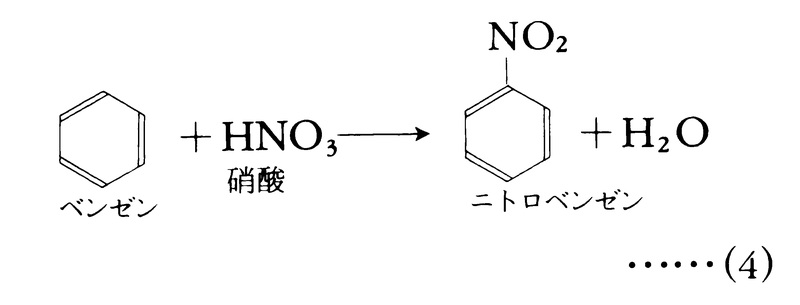
式(3)においてBとYがそれぞれ二つの有機反応物A:BとX:Yの小さな部分であり,その結果,生成物B:YがA:Xに比べて著しく低分子量(たとえば水)であるとき,この種の反応を縮合condensation反応と呼ぶことも多い。不飽和結合を含む反応物にもう一つの反応物が結合する場合,この種の反応を付加addition反応という。一般式では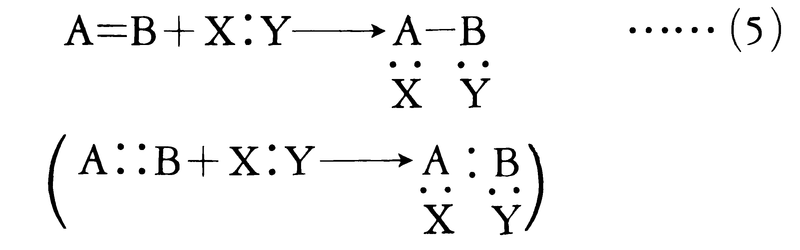
不飽和結合は二重結合でも三重結合でもよい。典型的な付加反応としてアルケンに対する臭素や水素の反応がある。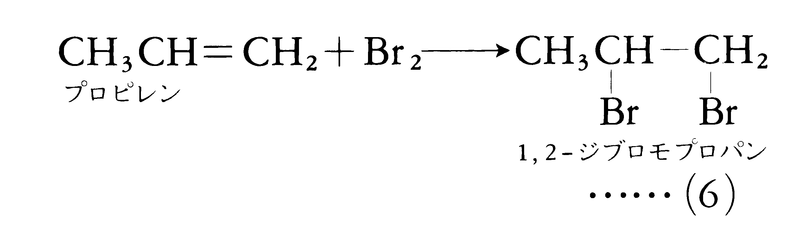
脱離elimination反応は形式的には付加の逆反応である。反応物の一部が脱離して不飽和化合物が生成する。一般式では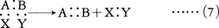
アルコールの脱水によるアルケンの生成はよく知られた脱離反応の例である。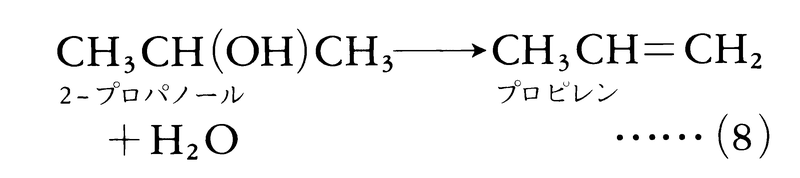
転位rearrangement反応では,反応物と生成物の分子式は等しいが,結合の切断と生成によって反応物の異性体が生じる反応である。一般式では
A:B:X─→X:A:B ……(9)
転位反応は単独に起こるよりも,他の反応(置換,付加,脱離のいずれでもよい)にともなって起こることが多い。たとえばベンゼンと塩化プロピルとの反応(フリーデル=クラフツ反応)においては,生成物はプロピルベンゼンではなく,転位をともなうイソプロピルベンゼンである。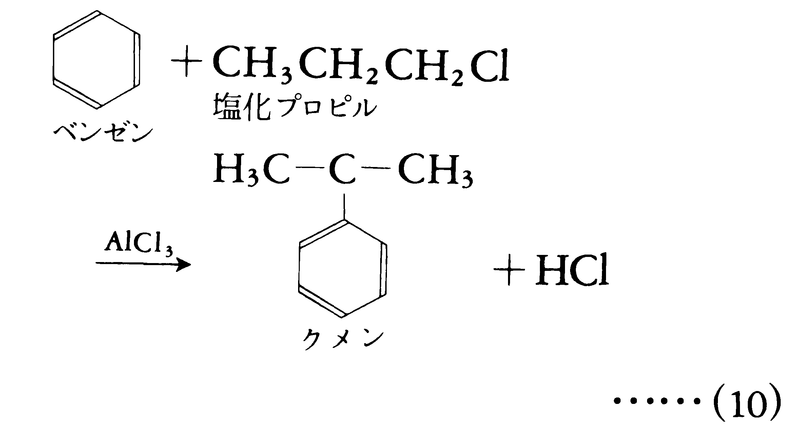
これは中間に生じたカルボカチオンCH3CH2CH2⁺がより安定な(CH3)2C⁺に転位し,その後置換反応が起こったためである。
反応機構
式(10)をみても,反応が置換反応の一種であることがわかるだけで,転位がどの段階で,どのような仕組みで起こったかを知ることはできない。一般に化学反応式は反応物と生成物の化学量論的関係を示すだけで,反応の詳細な経過を示してはいない。一般の化学反応と同様,有機化学反応も1段階ではなく多段階で進行する。たとえば反応
A+B─→C+D ……(11)
において,AとBはまずEを与え,これがFとGとなり,ついでFとGが反応してCとDを与えるものとする。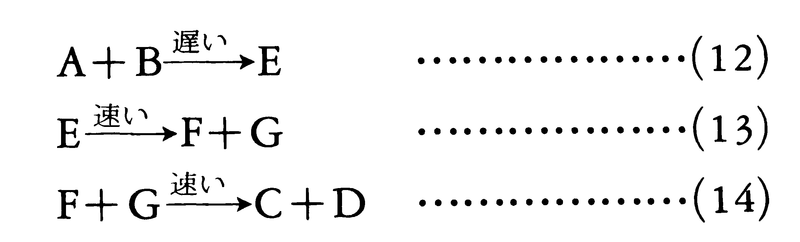
式(12)~(14)を反応(11)の反応機構という。反応の各段階は一般に異なる速度で進行するが,上の例では第1段階(式(12))の反応速度が遅く,全反応速度はこの段階によって規定されるので,反応の律速段階rate determining stepという。反応の経過に際して生じるE,F,Gが実際に単離可能である場合,これらは反応中間体という。E,F,Gが高い反応性のためごく短寿命で,その結果単離はできないがその存在を証明しうる場合,これらは遷移状態transition statesと呼ばれる。遷移状態にある系を活性錯体activated complexという。反応の進行は,横軸に反応の進行,縦軸に反応系のエネルギーをプロットしたエネルギープロフィル図energy profile diagramで模式的に示される。遷移状態はエネルギープロフィル図においてエネルギーの極大値に対応する一方,反応中間体は高エネルギー状態におけるエネルギー極小値に対応している(反応速度)。反応機構を決めるためには,反応の中間体ないし遷移状態を知って各段階を明らかにし,さらに各段階の反応速度を求める必要がある。反応機構と反応速度との関係に関連してとくに重要なのは反応次数である。式(12)において反応速度vの測定を行うと,反応物の構造や反応条件に応じて
v=k[A][B] ……(15)
すなわち反応速度が二つの反応物双方の濃度に比例する場合と,
v=k[A]またはv=k[B] ……(16)
すなわち反応物の一方の濃度にだけ依存する場合がある。これらは二分子反応,一分子反応と呼ばれる。反応速度は律速段階によって決まるから,二分子反応と一分子反応の区別は律速段階がどのようなものであるかを決めるのに役立つ。
求核反応と求電子反応
これまで式(11)において二つの反応物AとBをとくに区別しなかったが,多くの有機化学反応ではその一方を反応を受ける主体である基質substrate,他方を基質を攻撃する試薬reagentに分けて考えると便利である。反応(4)においては,ベンゼンが基質,硝酸が試薬となる。イオン反応において基質に電子を与えることのできる試薬を求核試薬nucleophilic reagent(求核剤nucleophileともいう)という。求核試薬は電子対を基質に与えることができる。代表的なものとして,OH⁻,Br⁻,I⁻,NH3,H2Oなどがある。カルバニオン,ヒドリドイオンH⁻も求核試薬である。これに対して本来電子不足性であり,基質から電子対を奪う試薬は求電子試薬electrophilic reagent(求電子剤electrophileともいう)と呼ばれる。代表的求電子試薬として,H⁺,NO2⁺,Br⁺,AlCl3,ZnCl2,BF3などがある。イオン反応は関与する試薬の種類によって,求核反応と求電子反応に大別できる。これを置換,脱離,付加の区別と組み合わせた分類が最も一般的である。
求核置換反応
求核試薬Nu:が正電荷を帯びた基質炭素原子を攻撃し,結合している原子または基と置換する反応を求核置換nucleophilic substitution反応という。追い出される原子または基を脱離基leaving group(L:と略記)という。脱離基は結合電子対とともに炭素から離れる一方,求核試薬が新しい電子対をつくる電子を供給する。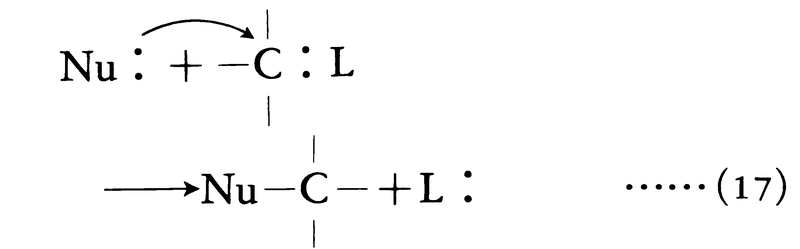
求核置換は電気陰性基が結合した脂肪族化合物に起こりやすい。たとえば臭化メチルと水酸化物イオンの反応を例にあげることができる。
CH3Br+OH⁻─→CH3OH+Br⁻ ……(18)
反応速度がv=k[CH3Br][OH⁻]で表されるので,この型の反応は二分子求核置換bimolecular nucleophilic substitution(SN2と略記)である。
この事実および基質が光学活性のときに反転(ワルデン反転)が起こることから,ヒューズE.D.HughesおよびインゴルドC.K.Ingoldらは,三角両錐形の五配位遷移状態を提案した。彼らの説によると,ワルデン反転は以下の反応機構で説明される。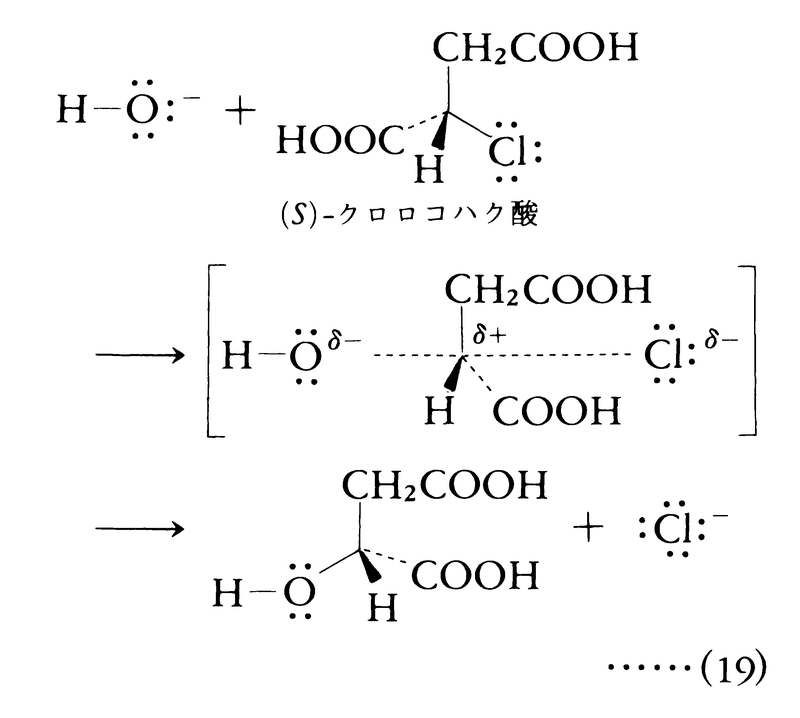
SN2反応の特色は基質の反応中心の立体化学的環境に著しく,たとえば同じ水酸化物イオンとの反応において,求核攻撃を受ける炭素が立体的に障害を受けている臭化第三ブチル(CH3)3CBrの反応速度はCH3Brの反応速度の1万分の1以下である。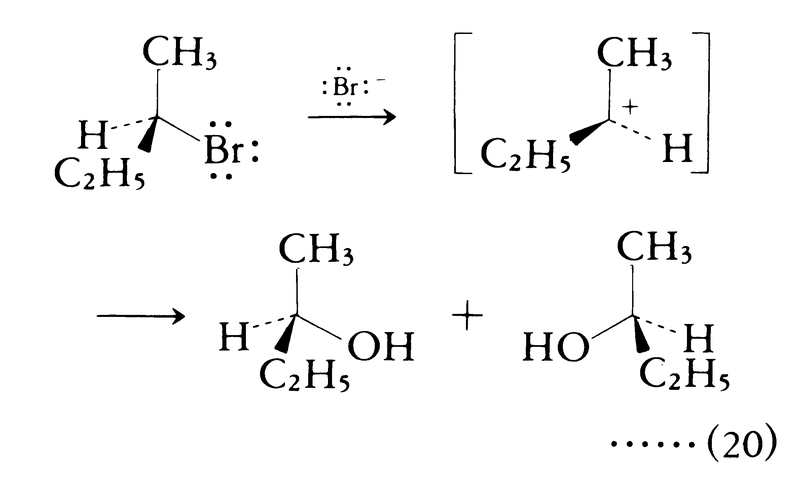
立体障害を受けている臭化第三ブチルのような化合物は,五配位遷移状態を経由しない別の反応経路をとる。この経路の反応速度は1次反応速度式に従う。また光学活性物質はラセミ化される。ヒューズおよびインゴルドの図式によるとこの反応経路は一分子求核置換unimolecular nucleophilic substitution(SN1と略記)であり,律速段階は三配位平面構造をもつ反応中間体カルボカチオンの生成である。
SN1反応の速度は律速段階で生成するカルボカチオンの安定性によって決まる。すなわち,より安定なカルボカチオンはより速く反応する。
芳香族求電子置換反応
電子過剰性のベンゼン環をもつ芳香族化合物は特定の場合のほかは求核反応を受けにくい。逆に求電子試薬E⁺がベンゼン環の水素原子と置換し,H⁺が脱離する芳香族求電子置換aromatic electrophilic substitution反応が最も一般的な反応となる。
ベンゼンのニトロ化,ハロゲン化,スルホン化,フリーデル=クラフツ反応などがこの反応に分類される。反応は多くの場合二分子反応で,律速段階はシクロペンタジエニルカチオン構造をもつ反応中間体の生成である。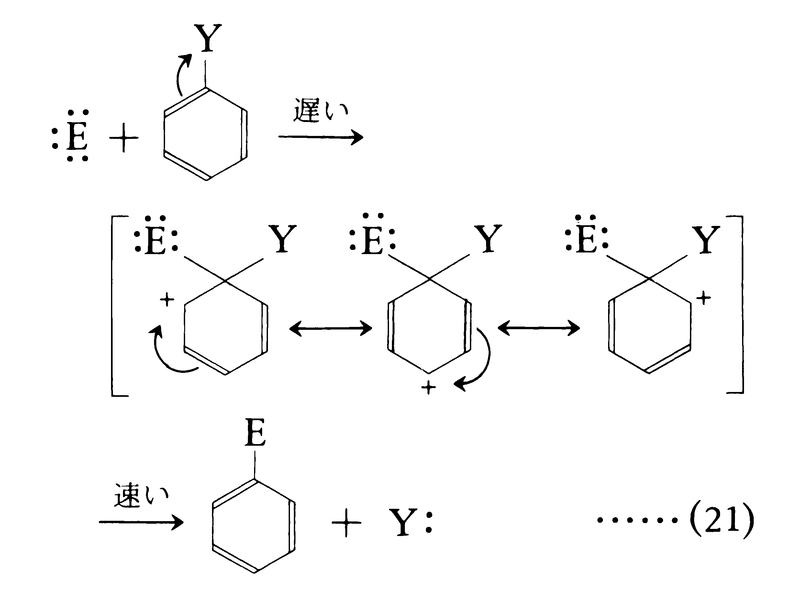
反応中間体は共鳴によって安定化される。置換基をもつベンゼン環に対する求電子置換は,新たに導入されるEがすでに環に存在する置換基とどのような位置関係になるかという配向orientationの問題と,ベンゼンに比べて反応性は高いか否か,という反応性の問題がある。これらはいわゆる置換基効果substituent effectとして知られている。
求電子付加反応
アルケンもベンゼンと同様電子過剰性であるから求電子攻撃を受けやすい。したがってアルケンと求電子試薬 との反応では,まず第1に多重結合に対する
との反応では,まず第1に多重結合に対する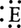 の付加が起こる。エチレンと臭素との反応では,まず求電子試薬ブロモニウムイオン
の付加が起こる。エチレンと臭素との反応では,まず求電子試薬ブロモニウムイオン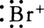 の二重結合への付加が起こる。
の二重結合への付加が起こる。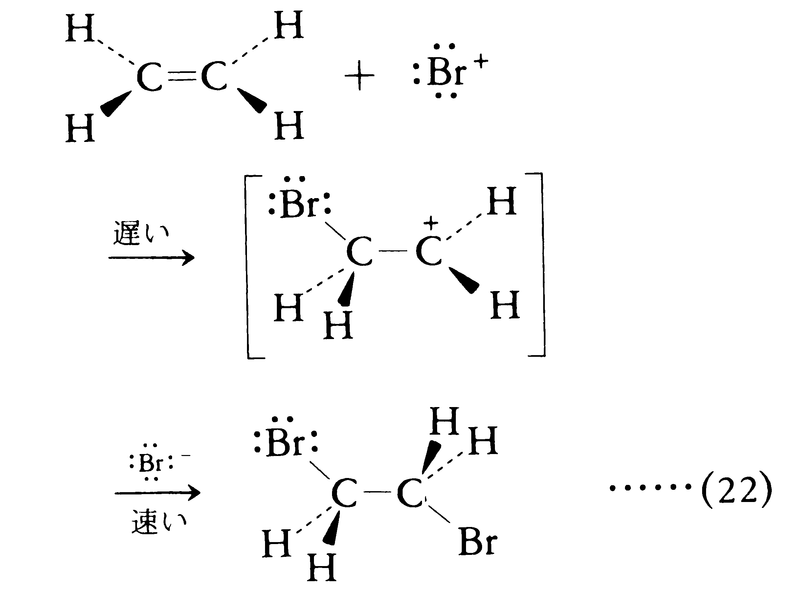
反応の第2段階は中間体カルボカチオンとE⁺の対イオン(この場合では臭化物イオンBr⁻)との反応である。構造上特別の制約がないかぎり,Br⁻は立体障害の少ない側すなわちBr⁺が接近したのと反対の方向から接近する。この種の付加の立体化学をトランス付加という。求電子付加に対してカルボカチオンでなくπ錯体を経由する機構も提出されている。
求核付加反応
炭素-炭素二重結合と異なり,炭素-酸素二重結合は分極しているため,同じ二重結合でも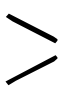 C=Oに対する付加は正電荷を帯びた炭素原子に対する求核試薬の攻撃が反応の原動力となる。多くの場合,この段階に続いて求電子試薬が酸素原子と結合する。
C=Oに対する付加は正電荷を帯びた炭素原子に対する求核試薬の攻撃が反応の原動力となる。多くの場合,この段階に続いて求電子試薬が酸素原子と結合する。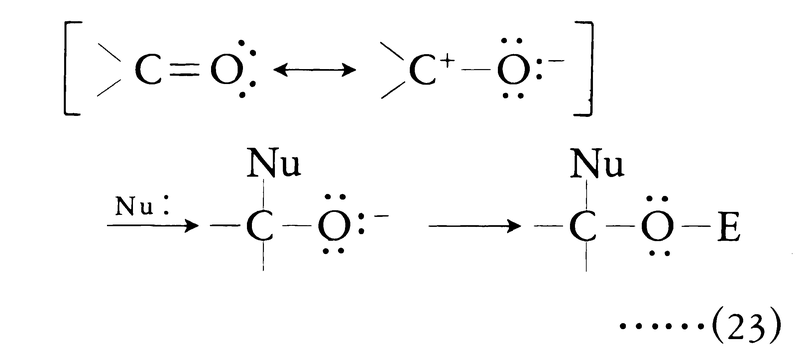
典型的な例は有機金属化合物(カルバニオンを生じる)とカルボニル化合物との反応によるアルコールの生成である(グリニャール反応)。このほか水素化リチウムアルミニウムLiAlH4などの金属水素化物によるカルボニル基の還元もこの反応に属する。
求核脱離反応
置換反応や付加反応と同様,脱離反応も種々の機構で起こりうる。典型的な脱離反応は求核置換と競争的に進む求核脱離である。求核試薬(多くの場合アルコキシドイオンや水酸化物イオン)が電気陰性基(脱離基でもある)によって活性化された水素を攻撃し,基質からプロトンとして引き抜く段階が反応の原動力である。プロトン引抜きと脱離基の脱離が同時に進行するのが二分子求核脱離(式(24)),脱離基の脱離が先行し,カルボカチオンが中間生成するのが一分子求核脱離(式(25))である。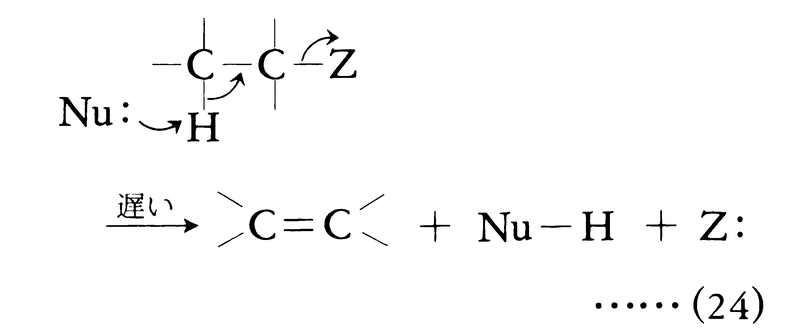
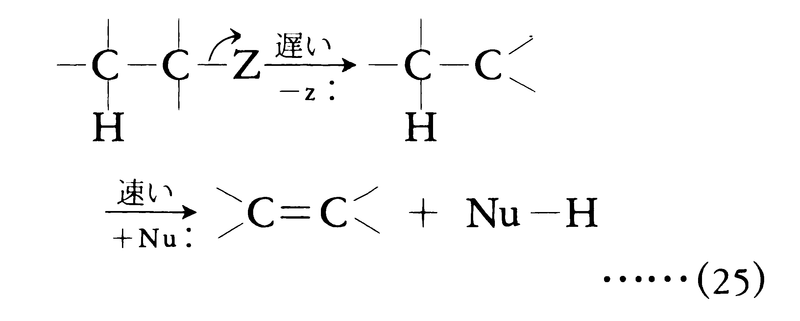
一般に引き抜かれる水素と脱離基とがトランス配座をとるとき,脱離は容易に進行する(トランス脱離)。
ラジカル反応もイオン反応と同様,反応の形式に応じて置換,付加などの区別を行うことができる。たとえばラジカル置換反応においてはラジカルX・が基質R-Hと反応して新しいラジカルR・を生じる。
X・+R-H─→X-H+R・ ……(26)
生じたR・がラジカルX・の発生源X2と直接反応してX・を生じるならば,最初に一度X・が生じればつぎつぎと式(26)(27)に示す反応が反復して起こることになる。
R・+X-X─→R-X+X・ ……(27)
このような反応の経過を連鎖反応chain reactionという。多くの場合,式(27)とは別の機構で最初のX・が生じる。ハロゲン分子X2の光開裂によるX・の生成はその一つである。
一方,2個のラジカルどうしが反応し,新しいラジカルが生成しないと,反応の連鎖はそこで切れてしまう。
X・+・R─→R-X
R・+・R─→R-R
X・+・X─→X-X ……(29)
連鎖反応の段階(式(28))またはこれに対応する反応を連鎖開始反応,反応(26)(27)の過程を連鎖移動反応,反応(29)の段階を連鎖停止反応という。連鎖反応の代表例としてメタンの光塩素化をあげることができる。連鎖移動の進行に応じてメタンの水素が順次塩素によって置換されていく。起こりうる諸反応の一部を示す。
移動 Cl・+CH4─→HCl+CH3・
CH3・+Cl2─→CH3Cl+Cl・
Cl・+CH3Cl─→CH2Cl・+HCl
CH2Cl・+Cl2─→CH2Cl2+Cl・
停止 CH3・+Cl・─→CH3Cl
CH3・+CH3・─→CH3CH3 ……(30)
ペリ環状反応
イオン反応,ラジカル反応のどちらにも属さない,特徴的なペリ環状反応pericyclic reaction(電子環状反応ともいう)と呼ばれる反応の一群がある。この反応では反応物の分子軌道どうしが相互作用して閉じたループをつくり,このループの中で電子対が再配列して結合の切断,生成が起こる。結合の開裂・生成は段階的でなく同時に起こる協奏反応であることが,ある反応がペリ環状反応であるための必要条件である。ペリ環状反応の最も一般的な例は,2種の不飽和化合物による環形成反応,いわゆる付加環化cycloadditionである。とくにジエンとアルケンとの反応によるシクロヘキセン環生成反応であるディールス=アルダー反応(ジエン合成)は名高い。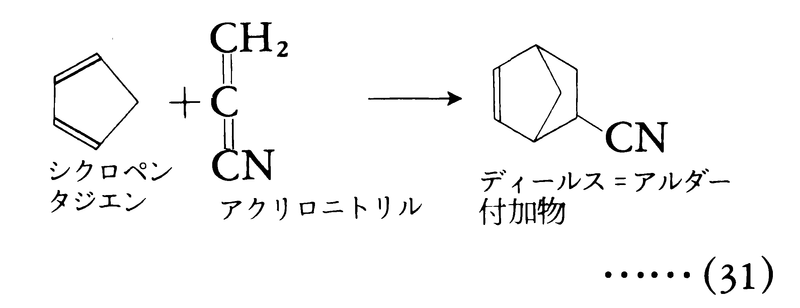
ディールス=アルダー反応をはじめとして,ペリ環状反応の例は古くから数多く知られていたにもかかわらず,これらを説明する統一的理論は1965年までなかった。この年,ウッドワードRobert Burns Woodward(1917-79)とホフマンRoald Hoffmann(1937- )は,電子軌道の対称性に基づいた新しい理論(軌道対称性理論またはウッドワード=ホフマン則)を発表した。これによって,理由が不明のままにされていた多くの有機化学反応の起りやすさの程度や,立体化学などが明らかになった。
人名反応
有機化学反応のなかでとくに合成に用いられるものは,その反応の発見者ないし,応用範囲や限界を明らかにした研究者の名前をつけて呼ばれ,人名反応name reactionと総称される。これらは反応機構や反応の型にはまったく関係ない。グリニャール反応,ディールス=アルダー反応などが人名反応の代表例である。これに対して,別の一群の反応はある特定の目的物質,あるいはそれの合成に必要な原料の名で呼ばれる。(フィッシャーの)インドール合成,マロン酸エステル合成などの例がある。
代表的有機化学反応
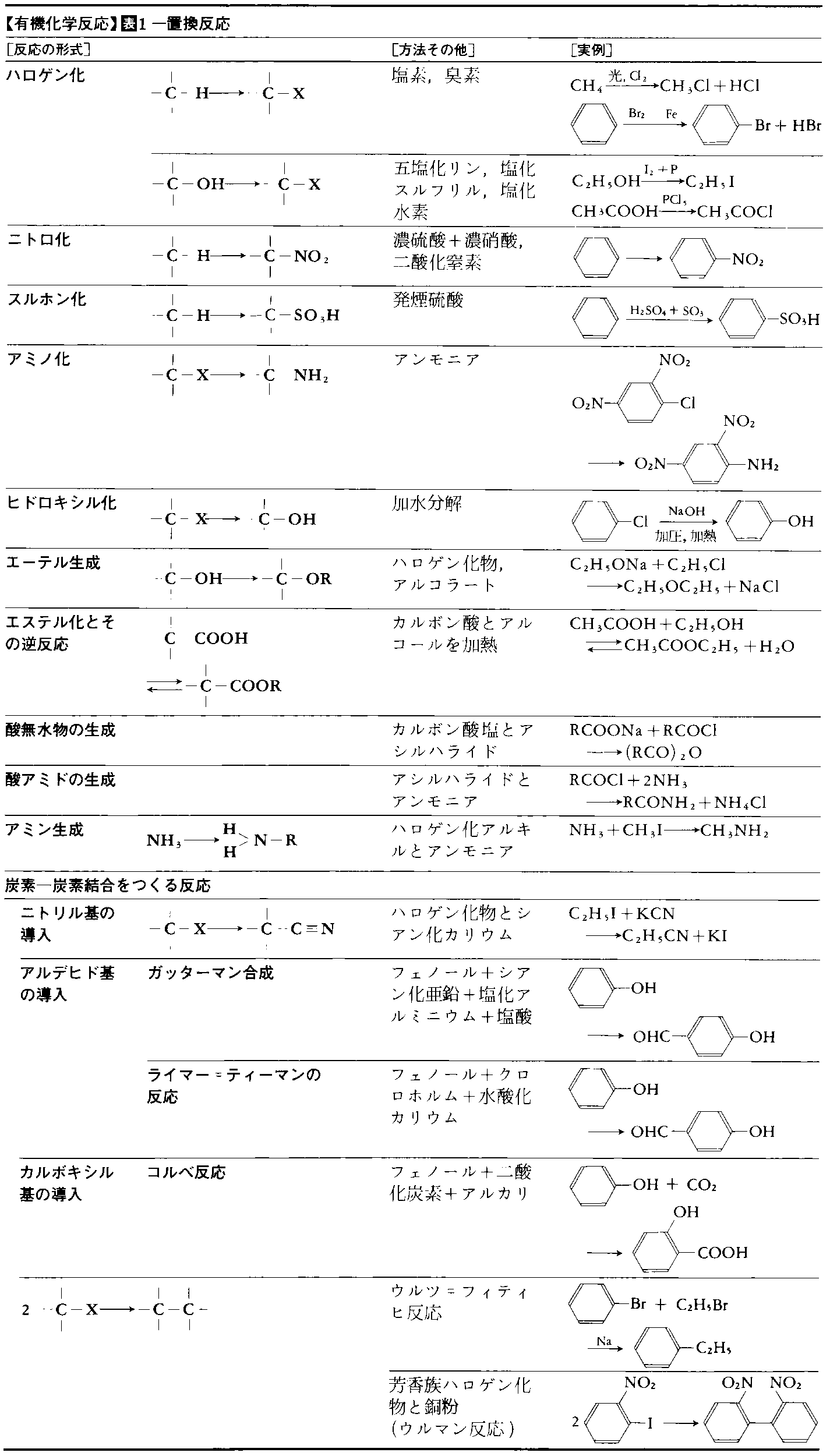
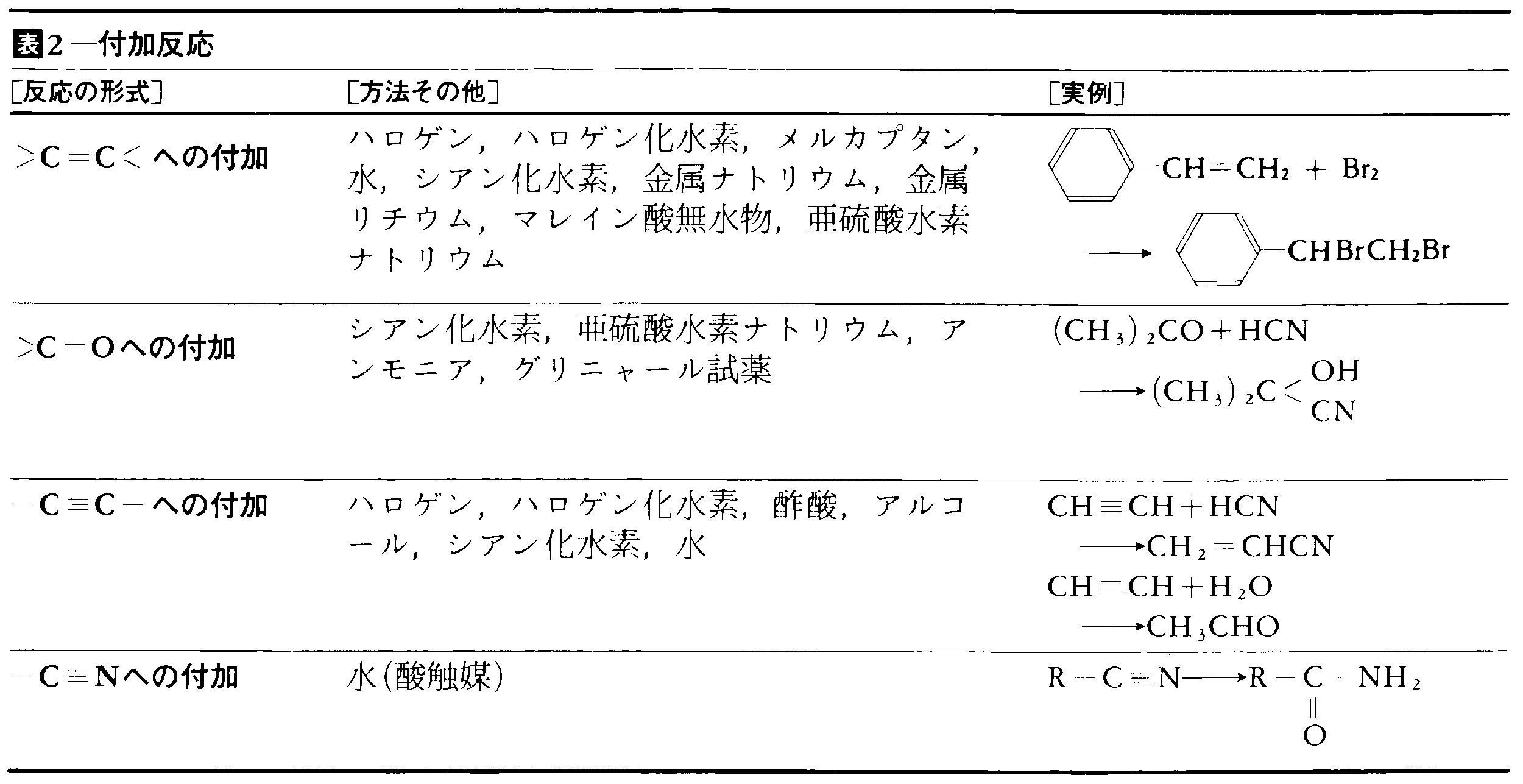
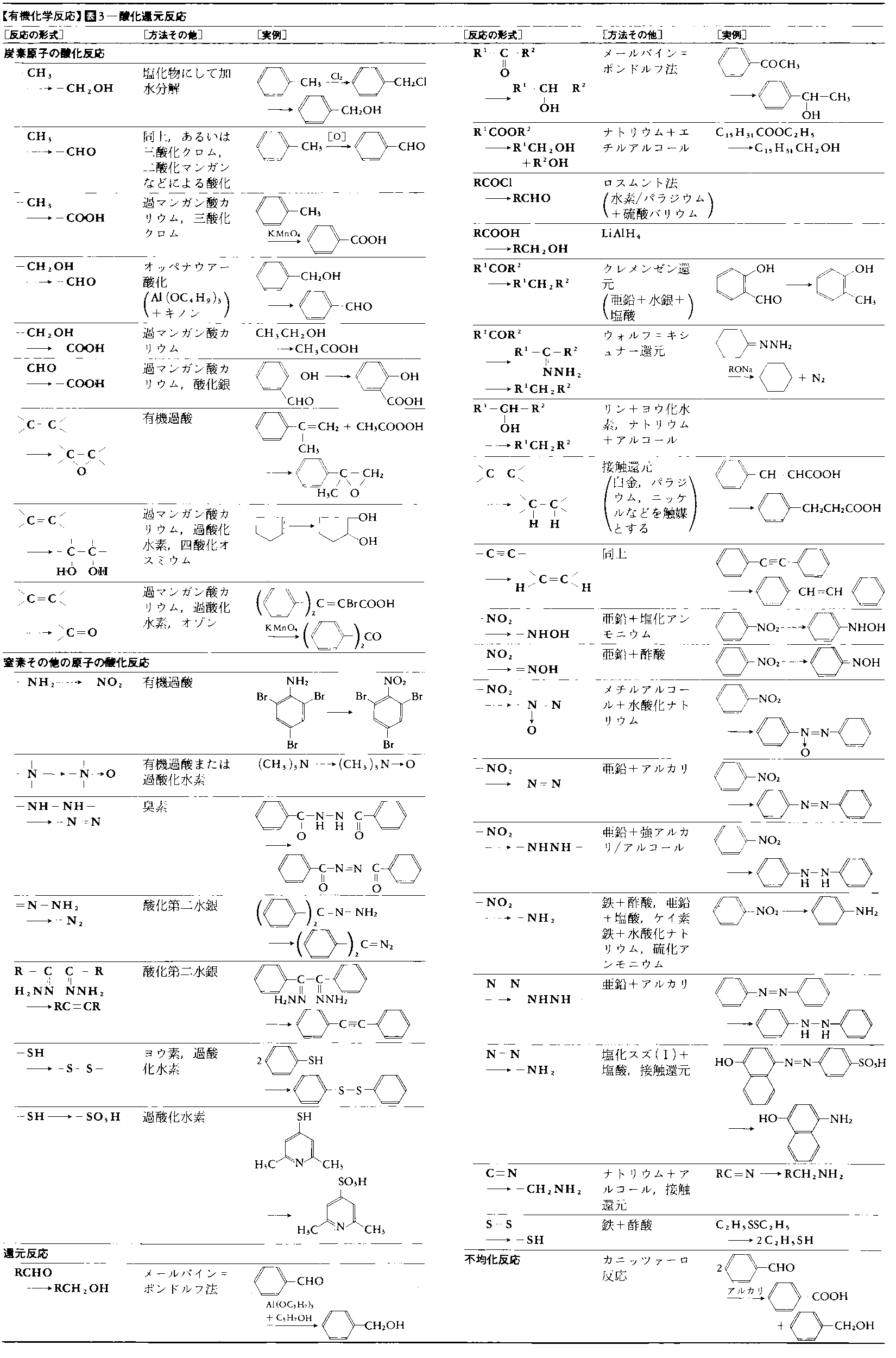
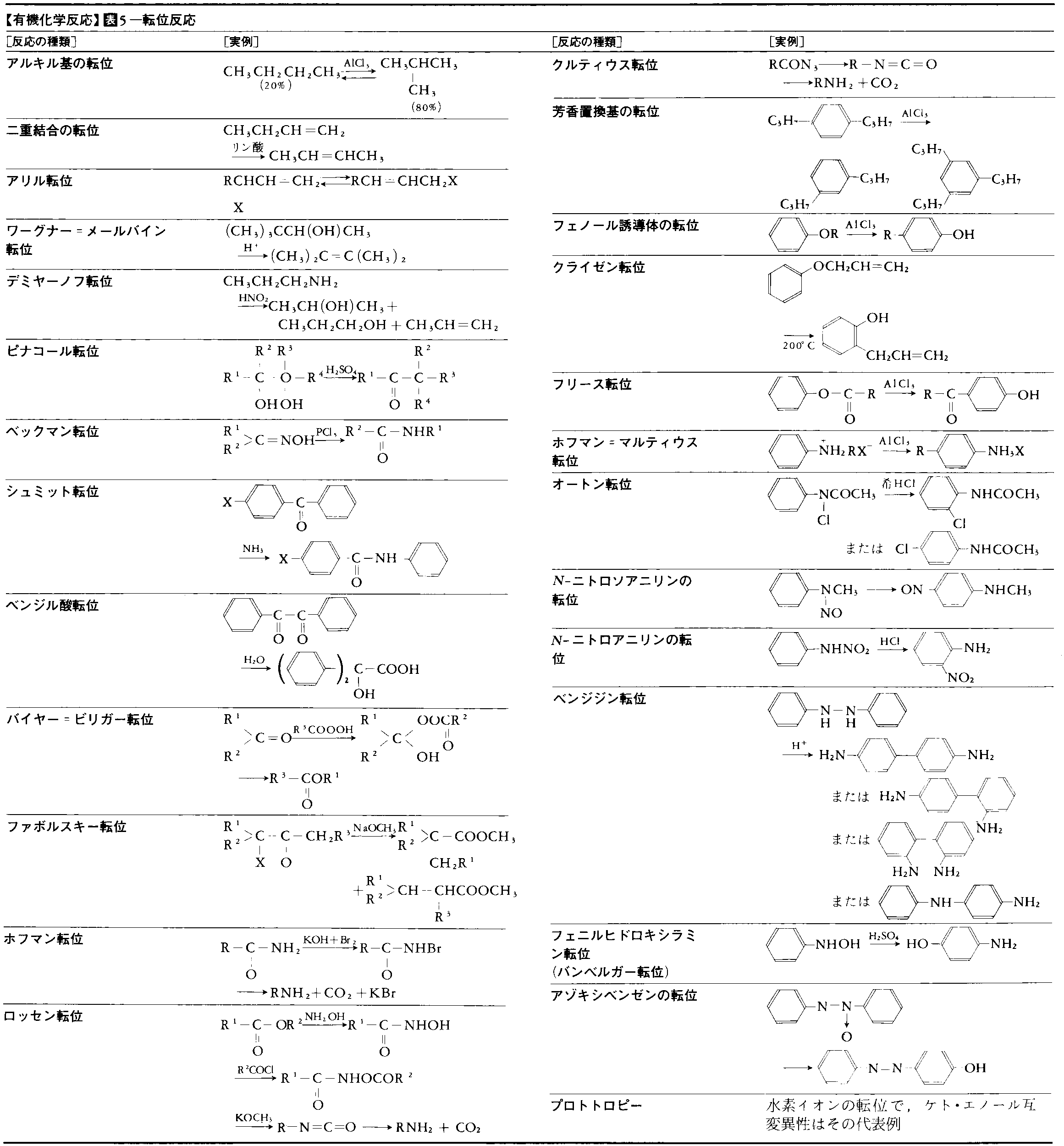
有機化学反応にはいろいろな分類のしかたがあり,実際にもそれらが併用されている。表1~5に主要な有機化学反応を表示した。
執筆者:竹内 敬人

出典 株式会社平凡社「改訂新版 世界大百科事典」改訂新版 世界大百科事典について 情報
Sponserd by ![]()