デジタル大辞泉 「医薬品」の意味・読み・例文・類語
Sponserd by ![]()
Sponserd by ![]()
出典 精選版 日本国語大辞典精選版 日本国語大辞典について 情報 | 凡例
Sponserd by ![]()
医療の用に供される薬品を医薬品とよぶ。一般に医薬品とはどのような性格をそなえているかをまず考えてみることにする。
医薬品は,純粋な化合物であれ,あるいは草根木皮のエキス等であれ,まず第1に〈物質〉である。すなわち,物理的,化学的にその性質を規定することができる物質である。たとえば一定の融点あるいは沸点,一定の溶解度,特有の旋光度等々物理化学的恒数をもつ化合物であるか,あるいはこれらの化合物の含有量を定められた混合物である。したがって医薬品は,一定の規格のもとに製造され,検定されうるものである。第2に,この〈物質〉が医薬品であるためには,人間の疾病の治癒,あるいは疾病にともなう苦痛を軽減するか,疾病の予防に役立つか等々に通ずる〈活性〉をもったものでなければならない。
さらに第3は,現代の資本主義社会においては,商品として流通している,すなわち商品として機能している物質である。この機能は,医薬品が原始的な型で生まれた太古の時代にも,物々交換の貴重な財貨としての特性をもっていた歴史を考えると,むしろ〈交換価値のある財貨〉としての性質をもつというべきであろう。
第4に,〈医の倫理〉は医師が行う医療に関して医師が守らなければならない〈義務〉を遂行することとして求められるのと同様に,医薬品を取り扱う責任を有する薬剤師が守るべき〈義務〉を含めて〈薬の倫理〉があることが,医薬品の特質となっている。
医薬品の発展の歴史を調べてみると,主として世界各地,各民族の食生活と密接に結びついて発展してきたことが明らかである。何万年,何千年の生活の知恵の集積が行われて,今日の医薬品となったものであるが,食物として採集して摂食した草根木皮が下剤の作用あるいは鎮痛作用があることを経験し,あるいは露出した鉱物の粉末が血止めの作用があることなどが経験され,やがてそれらの〈作用をもつ草根木皮,鉱物〉が意識的に採集され,そして,それらのものが,他部族との交流の際に有効な〈交換価値のある財貨〉と認められると,そこに作用をそこなわずに〈貯蔵〉するという技術が生まれたものと考えられる。そして永い年月の間に幾種も草根木皮や鉱物類の有効な組合せが経験から生み出され,伝承されてきたのである。それらの〈素朴な知恵〉が集積され,整理されてきたものが,前5000-前2400年のころにメソポタミア地方のシュメール人の手になったといわれる粘土板上に楔形文字で書き残された医薬品の記述に残されている。これには,250種以上の植物性の薬,180種以上の動物性の薬,120種以上の鉱物性の薬が含まれているといわれる。このことから,すでに治療を目的とした植物,動物,鉱物を含む医薬品の調製が行われていたことが推測される。ギリシア,ローマの古典医学は,一方では〈治療の論理〉を追究すると同時に,医薬品の種類,剤形を整備してきた。アラビアでは古代エジプトに興った錬金術が大胆に取り入れられ,その結果多種類の無機化合物が作り出され,これが大胆に医療の中に持ち込まれ医薬品の幅を広げた。また同時に技術的な進歩をもたらし,蒸留,昇華,溶解,灰吹法による金属の精錬,ガラス器具の発見,鉱酸類の製造などを生んだ。やがてルネサンス期を迎え科学と技術の乖離(かいり)が埋められたときに,華々しい科学の開花期を迎える準備が行われた。11世紀の十字軍のアラビア遠征,アラビアによる南ヨーロッパへの進攻などによる交流によって,アラビアの医薬学がヨーロッパに逆流した。ヨーロッパの各地にアラビアにならった新しい薬局が開設された。また11世紀にはギリシア・ローマ時代以来薬種を扱っていたピグメンタリウスの後継者が職人のギルトを模した〈調剤師同業組合〉(後の薬剤師同業組合)を結成し,錬金術によって発展したアラビア薬学の受け皿となり,親方による徒弟の徹底的錬金術教育が行われた。この同業組合に結集した薬剤師たちは,新たに医薬品の処方に組み込まれた医師の処方箋に従って調剤を行うと同時に植物分類学を発展させ,やがて分類学上の位置を確定した植物から,〈薬の精〉を導き出す仕事に手を染めた。18世紀には,これらの新しいエキス剤,あるいは単離精製された草根木皮の主成分が調剤の中に加えられるようになった。これが近世までの医薬品の製造の姿であった。18世紀に,蒸気機関が発明され,産業革命が起こると,個人の薬局の中で行われていた医薬品生産が,工場生産へと移行していったのである。
19世紀以後の化学,ことに近代有機化学の進歩と,R.フィルヒョー,L.パスツール,R.コッホ,C.ベルナールらによって打ち立てられた近代医学の進歩に支えられて,医薬品の製造が本格的な発展を示し,今日の医薬品製造へと進展してきたのである。
現代有機合成化学と,発酵学,製造工学が結びついて,医薬品の製造は化学工業の最先端部分となり,それによって生産される医薬品は,最も交換価値のある財貨の一つとして考えられている。
西洋の医学・薬学の体系が,古代バビロニアからギリシアの時代にかけて整理され,体系化されたギリシア医学に端を発しているのと同様に,東洋においても中国大陸では古代から各地の住民により,さまざまな経験医術が自然発生的につくり出されてきた。とくに中国大陸の広大な地域で,それぞれの地方の風土や生活様式に応じて,異なった医療方法が発達していたが,これらの地方医術は前8世紀から前3世紀にかけての春秋時代以降,各地方の統一,交流がすすむにしたがって,しだいに集成され,体系づけられていった。その代表的なものが,後漢(1~3世紀)のころの《神農本草》と,《傷寒論(雑病論)》である。前者は,西方山地に発達したとされる〈薬効ある自然物〉に関する知識をまとめたもので,中国医学における薬学(本草学)の基礎となったものであり,後者は,身のまわりに存在するありふれた薬物(生薬)を適宜に組み合わせて,その総合的効果が十分に発揮できる特定の条件の疾病に用いるという,当時の江南地方の医術における経験が整理され,一定の薬物を配合した処方に適応する条件(これを証という)という根本概念を把握し,体系化したものである。
漢方医学は,高度な臨床治療体系をもち,非常に実用的なものであり,観念的,神秘的な色彩のまったく認められない実践的医学体系であった。日本には奈良時代,平安時代にかけて,仏教とともに,隋・唐医学として伝えられ,室町時代になって独自の発展をとげ,江戸時代の日本の主流の医学となった。
明治の諸改革にあたり,医薬事制度もまた西欧の先進諸国のそれにならって改変され,西洋医学・薬学を修めたもののみに,医師,薬剤師の免許が与えられることとなった関係上,日本の治療医学の表面からは漢方医学,漢方薬が消失したかに思えるが,西洋医学を学び医師免許を受けて医師となった人々の中にも漢方医学を熱心に勉学し,あるいは漢方を信奉する一般の人々にも支えられて,今日でも漢方医学の火が伝えられ,現代ではむしろ,漢方ブームとさえいわれる現象まで起こっている。〈漢方薬〉の項にその詳細な記述を譲るが,〈医薬品を患者に適用する〉部面で漢方医学の方法論を活用すべきことが,多くの医学者,薬学者の間でも考えはじめられている。
→漢方薬
新しい医薬品は,社会的要請に支えられた,たゆみない研究活動と,周到な注意と,多額の資金によって生み出される。
現在製薬企業で実行されている新しい医薬品の手順のあらましをまとめると,次のように要約される。
合成された新化合物,天然物より単離された化合物の中から,〈薬の候補者〉となるものを選び出す,いわゆるスクリーニングの段階から,薬理作用の検討,試験規格・試験方法の検討,薬理(治療)効果の検討,一般毒性・特殊毒性の検討,初期および本格的臨床実験の段階を経て,〈疾病の治療に有効であり,かつ毒性の少ない〉ことが立証され,新薬として許可申請されたものについて,薬事審議会の審査の結果〈良〉と判定されたものが新薬として承認されるのである。以下にその手順の中のおもなものについて述べ,医薬品の創製の考え方と手順を解説する。
(1)スクリーニングscreening スクリーニングとはふるい分けをするという意味で,この場合は,数多くの化合物(天然のもの,合成されたものなど)の中から,〈特定の生物活性〉のある化合物をふるい分け,〈薬の候補者〉となるものを選び出す作業をいう。一般的には,数十の試験項目を選び,流れ作業的に,化合物群をテストして,どれかの項目のテストで好成績を収めた化合物を〈薬の候補者〉として拾い上げる〈ランダム・スクリーニング法〉と,特定の薬効をもつ化合物を拾い上げる〈特定スクリーニング法〉を組み合わせて行われる。このスクリーニングで拾い出された化合物の作用性は,スクリーニング法に用いられた系で使用された生物あるいは生物の器官の母集団の半数に作用が現れる量,すなわち半数作用量(ED50,50%effective dose)で表現される。
(2)急性毒性試験acute toxicity test スクリーニングで強い作用性,すなわちED50の値が小さい〈薬の候補者〉が拾い上げられると,次に急性毒性試験が行われる。せっかく強い作用性があっても,毒性があまりに強ければ薬となりえないからである。急性毒性は1回の投与で,動物に現れる致死性によって検定され,ふつう半数致死量(LD50,50%lethal dose)で表現される。LD50/ED50が大きな数で表されるものは,大量に与えても致死毒性が少なく,かつ少量で十分な作用を現しうる〈有力な薬の候補者〉となるのである。
(3)亜急性毒性試験と慢性毒性試験 薬物は患者に対し1回だけの投与という場合はほとんどなく,普通は一定の期間,連続または断続的に与えられる。そこで,次に亜急性毒性および慢性毒性の検査が行われる。亜急性毒性試験subacute toxicity testでは1種類以上の動物について,1ヵ月以上,慢性毒性試験chronic toxicity testでは3ヵ月以上(1ヵ月以上の連用が予想されるものでは6ヵ月以上)の連続投与によって現れる毒性を,死亡率,体重曲線,病理解剖学的検査,血液学的検査,臨床生化学的検査など,広範囲に検査する。実験動物としてはラット,ウサギ,イヌなどが使われ,慢性毒性試験では,対照群を含めて,最大安全量と中毒量が含まれる4段階の量でテストすることが要求されている。また実験結果のばらつきを少なくし,再現性のある結果を得るため,動物の選定や飼育状態などへの配慮が必要となる。
この試験の結果,低死亡率で対照群との差が少なく,非可逆的変化のないものが〈さらに有力な候補者〉となる。
(4)特殊薬性試験 かつて薬物は,〈投与された個体に有効で,低毒性であればよい〉と考えられていた。しかしサリドマイド事件を契機に,〈胎児への影響〉を十分に調べなければならないことが製薬技術者に認識されるようになった。このような点から行われる特殊毒性試験が催奇形性試験teratologenicityである。催奇形性試験は1962-63年に全世界的に検討され,その骨組みがつくられたが,日本でも65年5月に厚生省の通達(薬製第125号通達)によって,新薬について催奇形性試験を行うことが義務づけられた。さらに75年に,その試験期間が器官形成期から妊娠前~離乳期と拡大された。この通達では,動物の種や数(齧歯(げつし)類と非齧歯類各1種,計2種,一群約30匹,ウサギでは10匹),投与経路(実際に使用する経路)や期間,投与量などが規定されている。これらの規定によって投与したのち,胎児や産児の成長や発育の異常,症状の有無,生殖能力,母動物の次産児などを検査して,その薬物の催奇形性の有無が調べられるのである。
このほか特殊毒性検査に,発癌性試験(慢性毒性試験などで発癌性の疑われるものでは,ラットやマウスを用いて2年間または全生涯の連続投与試験),薬物依存性試験がある。
(5)臨床試験 上記の特殊毒性試験までを非臨床試験という。ここまでの試験で〈作用性〉と〈毒性〉に関して科学的評価に耐えられる結果を得てパスした〈薬の候補者〉は,次にヒトつまり患者に対して〈疾病の治療に有効であるか〉〈安全であるか〉がテストされる。
非臨床試験では,反応のばらつきを可能なかぎり極少化し,かつ再現性ある数値として把握するために,純系の,疾病のない動物を,一定の環境で飼育する,という3点に留意して試験が行われた。しかし臨床試験では,疾病をもつ,さまざまな条件にある患者に対しての検討であるため,非臨床試験での人為的な条件づくりはすべて適用できない。したがって,この臨床試験で当然出現する〈反応のばらつき〉をどう処理して,科学的評価に耐えられる結果となしうるかを考慮した実験計画が必須となる。現在は次の4段階の試験として行われている。
(a)第1相試験 少数の健康人の志願者に対して,主として安全性の確認を目的として行われる。ここでは,常用量と推定される量での副作用の有無やその内容が検討される。
(b)第2相試験 第1相試験によって健康人に対する安全性が確認されたものについて,少数の,適症と考えられる患者を対象に行われ,適応症,用法,用量,有効率,作用の発現,副作用などについて検討される。つまり,適応症に対する有効性と安全性が検討される。
(c)第3相試験 この試験には比較試験と対照を置かずに多数の患者を対象とした拡大臨床試験があり,前者には単純盲検試験single blind testと二重盲検試験double blind test(二重盲検法ともいう)がある。これらのうち,現在は二重盲検試験が最も科学的評価に耐えうる試験とされている。
二重盲検試験とは,参加した患者を,層別化し,無作為抽出によって2群に分け,コントローラーが保持した割付表に従って,一群には試験薬(真薬)を,他の群にはまったく有効性をもたない偽薬placeboあるいは標準治療薬active placeboを与える方法で,患者も管理する医師も,だれに真薬が投与されているかわからない,ということから二重盲検法と命名された。現在,医薬品の製造承認書に必要な資料として,5ヵ所以上,150例以上,1主要効能あたり,1ヵ所20例以上の臨床例が必要とされている。
(d)第4相試験 市販後の追跡試験で,日本では新開発医薬品については,6年間の副作用報告が義務づけられている。この段階で副作用が発見され,使用法が修正されることもある。薬のある種の副作用については,世界保健機関(WHO)を中心に国際協力体制がつくられている。
アメリカ食品医療品局(FDA)は,1963年,GMP(Good Manufacturing Practiceの略。〈医薬品の製造と品質管理に関する基準〉)を制度化した。WHOも69年にGMP勧告を出し,日本も73年に厚生省と日本製薬工業会がJGMPを作成し,76年から実施されている。GMPとは,医薬品の製造に関し,施設,設備,製造に従事する人の教育と組織についての基準を定めたもので,製造工程や製品の品質,衛生や安全性への厳しい管理が義務づけられている。さらに,GMPに続いて,GLP(Good Laboratory Practiceの略。〈非臨床の安全性試験に関する基準〉)が,79年アメリカで実施され,日本でも厚生省がこれを定め,82年から実施されている。これは非臨床試験での動物実験の建物や施設,設備や,その衛生と安全管理,さらに試験計画やデータの管理など,非臨床試験の成績と信頼性を高めるための基準である。このように,医薬品の創製と製造については厳しい制約が設けられている。88年,臨床試験に参加する患者の人権を守り,かつ科学的な試験結果を得ることを目的としてGCP(Good Clinical Practiceの略。〈医薬品の臨床試験の実施に関する基準〉)が制定,実施されるようになった。また,医薬品副作用情報の収集と周知徹底を図ることを目的として,やはり88年にGPMSP(Good Post Marketing Surveillance Practiceの略。〈医薬品の市販後調査の実施に関する基準〉)が制定された。
また88年,世界の主要製薬企業30社が〈医薬品が本来的に有するベネフィットとリスク,それに対する科学的評価,医薬品の使用環境としての社会システムの変化,社会に許容されるリスクの範囲,リスク規制の意思決定に影響を及ぼす科学的および政治的要因,医薬品の正しい理解を促進するための情報開発と伝達〉などを討議し,〈医薬品のリスクとベネフィットの評価-分析および対応策Risk/Benefit Assessment of Drug-Analysis & Response〉の頭文字をとったRAD-AR協議会を発足させた。
90年,日米欧の3極の間で〈医薬品の承認〉への手続きに関して,各国の規制を3極で共通化するための〈医薬品規制〉ハーモナイゼーション国際会議(ICH)〉が発足し,97年の会議までに4回の会合が行われ,3極における規制の共通化の大筋が合意される段階に達した。
ヒトを含め生物に薬を投与すると,それぞれの薬に対する反応性の違いのため,結果に〈ばらつき〉が生じる。このばらつきの主要な原因としては,(1)個体の遺伝的素因による変動,(2)疾病による変動,(3)衛生環境による変動,の3点が考えられる。そこで,非臨床試験では,純系の,疾病のない正常動物を,一定の環境下で飼育して検査するというように,反応のばらつきを小さくし,実験結果が科学的で再現性あるようにくふうされている。さらに臨床試験では,個体による反応のばらつきを無作為に均等化することによって,比較試験が行われる。
このように緻密(ちみつ)な試験が行われるにもかかわらず,薬害による不幸な事件が後を絶たない。そこで副作用とはなにかについて考えてみたい。
現在,医薬品の投与によって,患者にひき起こされる障害をすべて〈副作用〉という。このような障害(副作用)は,(1)薬の投与量,処方,交付の過誤,乱用,(2)患者の体質,たとえば過敏性体質,(3)長期使用,の三つに起因すると考えられる。このうち,(1)は医療過誤に属する問題であり,(3)では,副作用がある程度予測され,そのうえで無処置のために起こる障害との軽重で判断されて与えられている場合が多いと考えられるので,ここでは(2)を中心に副作用について考える。
(a)予想される〈望ましくない作用〉 薬の開発の非臨床,あるいは臨床試験の段階で,〈望ましくない作用〉が見いだされる場合がある。この場合,その作用を,〈その薬の薬理作用に固有の望ましくない作用〉と,〈副次的な望ましくない作用〉に分けることができる。前者の例としては,鬱血(うつけつ)性心不全に対して,かつてよく用いられたスルホンアミド型利尿剤が,その利尿の機構からみて,低カリウム血症をひき起こすことが考えられるような場合である。また後者の例としては,抗ヒスタミン剤によって眠気を催す例で,直接求められている薬理作用と関係はないが,経験的に知られているものである。この両者ともあらかじめ,この作用に対する処置を用意することができるので,処置しやすい副作用ということができる。
(b)予測しえない〈望ましくない作用〉 各種のテストで認められなかった〈望ましくない作用〉で,患者の特殊な肉体条件のため,普通では発現の確率が推定できないような〈望ましくない作用〉である。
生理学,生化学,薬理学の進歩によって,生体のしくみも細部にわたって解明されるようになってきたが,これらの実験的裏づけは実験動物によって得られたもので,ヒトについての実証的研究でできないことから,ヒトについては動物実験から類推する以外にない。今後,さらに多くの因子についての知識が増えれば〈予測しえない望ましくない作用〉は減少すると期待されるが,こうした間接的な方法をとっていることから,ヒトについて,〈望ましくない作用〉が発現することがある。ヒトの生理に関した〈望ましくない例〉としては次のようなものがある。
代謝の影響(ヒトに毒物が与えられると,生体内ではUDP-グルクロン酸転移酵素によって,毒物はグルクロン酸抱合体となって尿中に排出され解毒される。ところが,肝臓疾患などによってこの機構に欠陥が生じると,〈予測しえない望ましくない作用〉が生じると考えられる),環境の影響(たとえば,食習慣によって,ビタミン類や微量元素が欠乏し,十分に細胞の酵素の代謝能が機能しないために,〈望ましくない作用〉が発現する),医薬品の併用(現在,薬理学者が科学的に解析できるのは2種の薬物の相互作用までであり,3種以上の相互作用の解析は不可能である。したがって,相乗作用または拮抗作用によって,予想しえない反応が起こる場合が考えられる)。また個人に特有な場合としては,遺伝的素因(遺伝的酵素欠損など),免疫感作(ペニシリンショックなどがこの例で,症状自体はその薬の固有の薬理作用とはなんの関係もない。このような反応は,患者の体液あるいは細胞内に抗体が存在するか否かによる),薬物依存と薬物嗜癖(向精神薬などに重要)などがある。
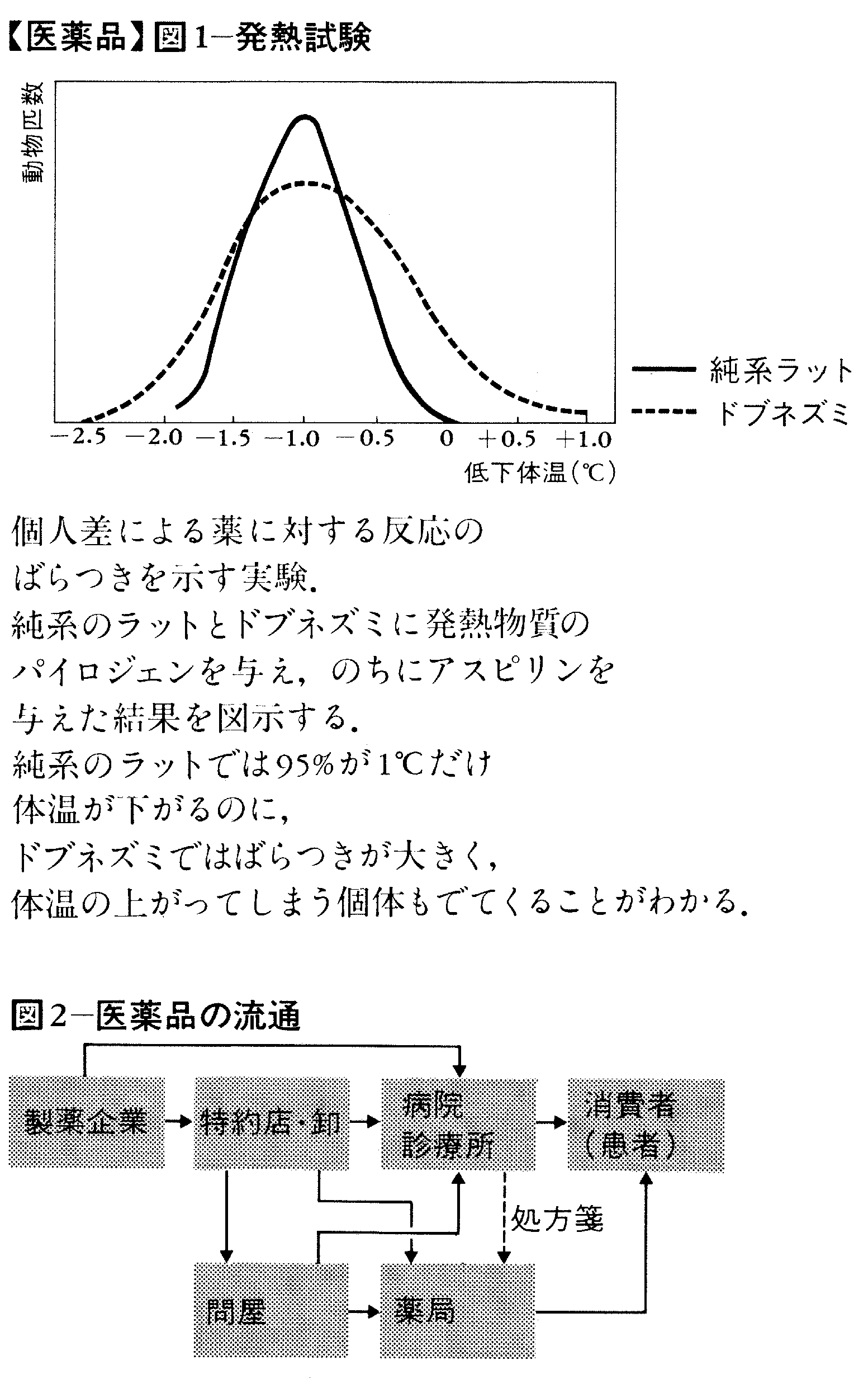
(c)個人差による反応のばらつきとはどのようなものか 実際に報告された例として,本来解熱効果をもつアスピリンを投与された子どもで,逆に発熱する人があった。なぜこのようなことが起こるのか。
そこで次のような実験を行ってみる。あらかじめ発熱物質のパイロジェンを与えて発熱させた純系のラットにアスピリンを与える。その結果,図1のような正規分布を得たとする。つまり,95%以上が1℃だけ体温が下がることになる。同じような実験を純系でないドブネズミ(病気をもつか否かにかかわらず)に対して行って,プロットして,図1のような結果を得たとすれば,アスピリンによって,3℃以上も体温が低くなるものから,逆に体温が上がってしまうものまで出る,ばらついた結果となることがわかる。
また薬物を患者に与えたとき,その薬物が有効に作用するためには,一定以上の濃度の薬物が血中に保たれる必要があると考えられている。ふつう薬物を投与されると,徐々に血中濃度は上昇し,一定時間後に減少していく。そこで,有効性をもたせるために,薬物の血中濃度はある一定時間,有効濃度以上で,しかも中毒量以下にあるようにくふうがなされる。動物実験では,1本の線で示しうるほどばらつきを少なくすることができるが,ヒトでは,ある幅が出,場合によっては,中毒量を超える人が出ても不思議はない。
以上のように〈ばらつき〉とは,個々の肉体的条件の差から反応性の差が生ずることである。薬の創製,製造にあたっては,あらかじめ,このばらつきを前提にして,各種の試験が行われるが,ばらつきの幅が,ある範囲を越えて出現すると〈予測されない望ましくない作用〉が出現することになるのである。
医薬品は人の生命,健康に直接かかわるため,日本をはじめ各国政府は,その流通に対して種々の法律を定めている。
医薬品に対する法規制は,1870年(明治3)の売薬取締規則にはじまり,売薬規則,薬品取扱規則等を経て,89年,薬品営業並薬品取扱規則(薬律と呼ばれている)にまとめられ,薬事制度の根幹となった。これらは,1943年薬事法(旧々法)が制定され,以前の3法が一つにまとめられた。このときから,医薬品製造業は主務大臣の許可を必要とするようになった。60年の大改正により薬剤師法と薬事法が分離され今日に至っているが,79年新薬事法が成立し,品質とともに有効性と安全性の確保が規定された。これにより薬剤師は必然的に〈患者に適用する医薬品の品質,有効性,安全性について責任を持つ〉ことが要求されるようになった。さらに93年の改正によって〈国民の医薬品へのニーズの多様化にこたえる医薬品の迅速・適格な研究開発と供給〉が求められた。同時に,薬剤師法によって患者への医薬品説明の義務が課せられた。また従来はおもに医療施設や医師を中心とした規定であった医療法が1985年,92年に改正され,薬局が医療施設として規定され,薬剤師が医療の担い手として明記され,地域医療の一端を担う医療人として規定された。
現在日本の医薬品の流通は図2に示した経路に従って製薬企業から消費へ伝達されている。製薬企業で生産される医薬品のうち約80%に相当するのは医家向け医療用医薬品prescription drugsで,特約店や卸を経て,あるいは製薬企業から直接病院診療に直結する経路で供給される。病院・診療所より処方箋が発行され,それに従って薬局(調剤薬局)から医療用医薬品が消費者(患者)に渡される,いわゆる医薬分業形態の流通にのる部分は10%以下である。残りの20%弱の医薬品は一般用医薬品OTC drugs(over the counter drugsの略)である。
卸,問屋も,医療用医薬品と一般用医薬品とを専門に扱う二つの系統に分かれている。小売業種も,薬局,薬店(薬種商),一般販売業,特例販売業の4種に分かれ,薬局では薬剤師が処方調剤や自家製剤を行い,指定医薬品(厚生大臣が品目を指定し,取扱い上の規制をしているもの),要指示薬品(乱用による弊害のゆえに,とくに取扱いを厳重にしているもの,および医師の処方箋または指示によらなければ交付を禁じられている医薬品)等,医薬品全品目を販売することが認められている。一般販売業では,薬剤師を専任管理者とする処方調剤などは行えない。薬種商(薬店)は,一定の資格をもつが,調製剤を行えず,指定医薬品の販売ができない。特例販売業は,品目と地域が限定されていて,営業規模はきわめて小さい。
そのほか,配置薬業と呼ばれる独特な流通が現在でも5%弱を占めているが,これは,特定の製薬企業が一般大衆薬の基準以内の医薬品を,消費者家庭に1ヵ年配置し,使用した医薬品の代金を翌年度に回収する制度で,富山,和歌山を中心に,この流通機構をもつ企業が存在する。
医薬品にはどのようなものが含まれるのであろうか。一般的には鎮痛剤,解熱剤,強心剤等々呼びならわされているが,薬事法には第2条に医薬品を次の各号に掲げる物をいうとして,(1)日本薬局方に収められているもの,(2)人または動物の疾病の診断,治療または予防に使用されることを目的とされているものであって,器具器械(歯科材料,医療用品および衛生用品を含む)でないもの(医薬部外品を除く),(3)人または動物の身体の構造または機能に影響を及ぼすことが目的とされているものであって,器具器械でないもの(医薬部外品および化粧品を除く)となっており,その基原,薬効,使用目的,薬事法などにより分類される。
基原によって分類すれば,化学薬品,生薬,油脂,蠟,精油類,生物学的製剤,血液製剤,抗生物質,放射性医薬品となる。また多くの医薬品は製剤に加工して使用されるため,製剤を剤形によって散剤,錠剤,軟膏剤,注射剤,エキス剤等に分類されており,日本薬局方の製剤総則には25の製剤が収録されている。適用法のうえからは,内服薬,外用薬,注射薬のように分類され,使用目的から,診断薬,治療薬,予防薬があり,また使用区分からは,医療用医薬品,一般用医薬品の区別がある。
薬効からの分類として実用的に繁用されているものに,日本標準商品分類がある。これに対して,国際十進分類Universal Decimal Classification(UDC)は理論的に扱われていると評価されており,その615.2として主作用による医薬品の分類が記されている。
しかし,医薬品の創製の項にも記載したとおり,〈薬〉は,合成化合物,天然の生薬の成分,動物組織の成分,微生物の代謝産物等基原を異にするさまざまな化合物の中から,スクリーニングにより〈薬の候補者〉を選び出し,毒性,薬理作用,安全性を動物実験によって科学的に把握し,さらにこの結果に基づいてヒトについて安全性,疾病および疾病にともなう諸症状を軽減する能力を検討したうえで誕生する。この過程をふり返ってみると,医薬品の開発の段階で認められた〈化合物としての生物活性〉と〈薬としての治療効果〉は必ずしも同義でないことは明らかである。たとえUDCにおけるがごとく,主作用による分類が合理的であるとしても,たとえばアスピリンは,周知のように主作用は解熱鎮痛であるが,またこの医薬品は脳血栓の予防剤として広く使用される薬物である。このことを考えると医薬品の機械的な分類はあまり意味のあるものとは考えられない。
→薬
執筆者:辰野 高司
出典 株式会社平凡社「改訂新版 世界大百科事典」改訂新版 世界大百科事典について 情報
Sponserd by ![]()
医療に用いる薬品をいう。医薬品医療機器等法(旧、薬事法。昭和35年法律145号)の定義によると
(1)日本薬局方に収められているもの
(2)人または動物の疾病の診断、治療または予防に使用されることが目的とされているものであって、機械器具、歯科材料、医療用品および衛生用品、プログラムおよびこれを記録した記録媒体でないもの(医薬部外品および再生医療等製品を除く)
(3)人または動物の身体の構造または機能に影響を及ぼすことが目的とされているものであって、機械器具等ではないもの(医薬部外品、化粧品および再生医療等製品を除く)
をさしている。なお医薬部外品とは、
(1)吐き気その他の不快感または口臭もしくは体臭の防止
(2)あせも、ただれなどの防止
(3)脱毛の防止、育毛または除毛
(4)人または動物の保健のためにするネズミ、ハエ、カ、ノミなどの防除
の目的に使用されるもののうち、人体に対する作用が緩和なもので、機械器具でないもの、およびこれらに準ずるもので厚生労働大臣の指定するものをいう。
医薬品には、医師や歯科医師によって使用されるか、またはこれらの者の処方箋(しょほうせん)、もしくは指示によって使用される医療用医薬品と、医師や歯科医師の処方箋なしで、薬局や薬店、通信販売、インターネットで直接購入できる一般用医薬品(大衆薬)、薬局・薬店で薬剤師の指導を受けて購入する要指導医薬品とがある。一般用医薬品は、医薬品医療機器等法により第一類医薬品、第二類医薬品、第三類医薬品に区分されている。第一類医薬品は薬剤師、第二類医薬品および第三類医薬品は薬剤師または登録販売者が販売に従事する。
[幸保文治]
出典 小学館 日本大百科全書(ニッポニカ)日本大百科全書(ニッポニカ)について 情報 | 凡例
Sponserd by ![]()
出典 ブリタニカ国際大百科事典 小項目事典ブリタニカ国際大百科事典 小項目事典について 情報
Sponserd by ![]()
Sponserd by ![]()
…一般にわれわれが〈くすり〉という言葉を口にしたり耳にしたりした場合,まず頭に思い浮かべるものは医薬品であろう。しかし,〈薬〉と同義である〈薬品〉という文字をみた場合,もちろん〈医薬品〉は頭に浮かぶが,それと同時に〈化学薬品〉を思い浮かべる。 化学薬品は,酸類,塩基類をはじめとする基礎化学工業製品,石炭タール成分,石油成分,あるいはそれらの分解産物,重合体など近代化学工業製品群を含んでおり,現代文明を支える化学工業製品の原料から最終製品にいたるさまざまなものが含まれている。…
…植物,動物および鉱物の天産物をそのままか,またはその一部,あるいは動植物のエキス,分泌物,細胞内含有物を乾燥など簡単に加工を施し,薬用に供するものである。しかし,直接医薬品となるものばかりでなく,生薬製剤,成分製剤の原料となるもの,ならびに香辛料,香粧品,工業薬品などの原料をも含む。生薬は乾燥などによって腐敗やカビなどの繁殖を防ぎ,動植物自体の酵素反応を妨げ,化学的,生化学的に経時変化の少ないものが常時利用できるようになった。…
…本来は薬物治療において主作用と対比される概念。医薬品の生体に対する最も著しい作用で,治療の目的に利用されるものを主作用とし,治療上不必要なもの,または障害となるような作用を副作用とする考え方が一般的であった。しかし最近では,むしろ患者側に立った見方から,医薬品の使用により生体に生じた有害な反応すべてを含むことばとして用いられることが多い。…
…健康保険法にもとづく保険制度の範囲で用いられる医薬品。通常,医薬品を服用する場合,薬店,薬局から購入して服用するか,医療機関で医師の診察を受けた後,投与されたものを服用する。…
…草本のときには薬草という。広義には古代から経験的に病気の治療および予防に用いられてきたもののほかに,医薬品の原料となるもの,香辛料,嗜好品,薫香料,香粧品や,未開社会において食糧を得るための矢毒や魚毒なども含まれる。したがって薬用植物とは人間および動物に対して,特殊な生理作用を有する植物ということもできる。…
…医薬品を,疾患の診断,予防ならびに治療の目的で,ヒトに適用する際の容量をいう。実験動物などに各種の試験をする場合にも同じ表現をとるが,この場合には必ずしも上記の目的のためとは限らない。…
…重要な医薬品について,その性状,品質,製法その他の基準を定めたもので,一般に法的強制力をもつ公定書のことを薬局方,略して局方という。薬局方の英語はラテン語のpharmacopoeaにさかのぼり,その語源はギリシア語のpharmakon(薬)とpoiia(作り方)の組合せに由来する。…
※「医薬品」について言及している用語解説の一部を掲載しています。
出典|株式会社平凡社「世界大百科事典(旧版)」
Sponserd by ![]()
福岡県福岡市博多区の櫛田神社の夏祭り。壮麗な山笠で知られる。今日,山笠には飾り山笠と舁き山笠(かきやまがさ)の 2種類がある。明治時代に電線が架設されて以降,物語場面の人形などを飾りつけた高さ 15m...